هموفیلی A چیست؟
هموفیلی A یک اختلال وراثتی وابسته به X مغلوب است که در اثر نقص در فاکتور VIII یا FVIII حاصل می شود. فاکتور VIII، فاکتور انعقادی عملکردی پلاسما میباشد. این اختلال در تعداد قابل توجهی از بیماران، نتیجه وقوع جهش ژنتیکی جدید و یا نوعی فرایند ایمونولوژیک اکتسابی است.
حالات بیماری و مرگ ناشی از هموفیلی عمدتا به دلیل هموراژ رخ میدهند. البته در گذشته بیماریهای عفونی مانند عفونت HIV و هپاتیت، به ویژه در بیمارانی که تا پیش از سال ۱۹۸۵فرآوردههای خونی دریافت میکردند، شیوع بالایی داشت.
بررسیهای آزمایشگاهی صورت گرفته روی افرادی که مشکوک به ابتلا به هموفیلی هستند، شامل شمارش کامل سلولهای خونی، بررسی فرایندهای انعقادی و سنجش فاکتور VIII است. در بیمارانی که ابتلایشان به هموفیلی تایید شده است، ارزیابیهای دورهای آزمایشگاهی شامل بررسی حضور مهارکننده فاکتور VIII و نیز غربالگری از لحاظ بیماریهای منتقلشونده از طریق انتقال خون و بیماریهای مسری مانند هپاتیت و عفونت HIV میباشد. اهمیت سنجش سطح فاکتور VIII ، در مانتیورینگ درمان جایگزینی آن است.
روند درمان هموفیلی شامل پروفیلاکسی، مدیریت خونریزیهای ایجادشده، القای تحمل ایمنی در بیماران دارای مهارکننده فاکتور VIII و درمان و توانمندسازی بیماران مبتلا به سینوویت ناشی از هموفیلی است. ارائه درمان به مبتلایان، از طریق مراکز جامع مراقبت هموفیلی صورت میگیرد.
دستهبندی بیماری
دستهبندی هموفیلی از لحاظ شدت، بر مبنای نشانههای بالینی حاکی از خونریزی و یا سطح ترکیبات پیشانعقادی پلاسما صورت میگیرد. مورد دوم، دارای کاربرد بیشتری است و طبقهبندی انجامگرفته بر اساس آن به شرح زیر میباشد:
- هموفیلی شدید: سطح فاکتور ۸ کمتر از یک درصد حالت نرمال است (< 0.01 IU/mL).
- هموفیلی متوسط: سطح فاکتور ۸ بین ۱-۵ درصد حالت نرمال است (01-0.05 IU/mL).
- هموفیلی خفیف: سطح فاکتور ۸ بیش از ۵ درصد و زیر ۴۰ درصد حالت نرمال است (>0.05 to < 0.40 IU/mL).
شکل شدید بیماری در کودکان زیر یک سال ظاهر میشود و ۴۳ تا ۷۰ درصد موارد هموفیلی A را تشکیل میدهد. شکل ملایم بیماری در کودکان یک تا دو ساله نمایان میشود و ۱۵ تا ۲۶ درصد موارد را شامل میشود. بیماری خفیف نیز در کودکان بالای دو سال رخ داده و ۱۵ تا ۳۱ درصد مبتلایان را درگیر میکند.
بررسی بالینی نشانههای خونریزی نیز به این علت استفاده میشود که بیماران با سطح فاکتور VIII کمتر از یک درصد، گاها فاقد خونریزیهای خودبهخودی بوده و یا دارای مقادیر اندک آن هستند؛ در نتیجه در بررسیهای بالینی به نظر میرسد که مبتلا به هموفیلی متوسط و یا خفیف باشند. عکس این موضوع در بیمارانی که فعالیت ترکیبات پیشانعقادی در آنها بین یک تا ۵ درصد است نیز صادق است و این افراد ممکن است از لحاظ بالینی علائم شدیدی داشته باشند.
پاتوفیزیولوژی هموفیلی A
تولید فاکتور VIII، پردازش و ساختار آن
تصور میشود که نواحی اصلی تولید فاکتور VIII در اندوتلیوم عروقی کبد و سیستم رتیکولواندوتلیال قرار داشته باشند. پیوند کبد، نقص فاکتور VIII در افراد مبتلا به هموفیلی را برطرف میکند.
وجود مولکولهای mRNA مربوط به فاکتور VIII در کبد، طحال و سایر بافتها تشخیص داده شده است. مطالعه تولید فاکتور VIII در ردههای سلولی آلوده شده (transfected) نشانگر این موضوع بوده که این ماده پس از تولید، وارد لومن شبکه اندوپلاسمی شده و در آنجا به پروتئینهای متعددی متصل میگردد. این پروتئینها، در تنظیم ترشح فاکتور VIII دخیل هستند و از جمله آنها، پروتئین متصل شونده به ایمونوگلوبولین است. تفکیک این دو از یکدیگر، فرایندی وابسته به انرژی میباشد.
شکسته شدن پپتید سیگنالی فاکتور VIII و افزوده شدن الیگوساکاریدها به آن در شبکه اندوپلاسمی به وقوع میپیوندد. هر دو پروتئین کلنکسین و کلرتیکولین که از چاپرونها هستند، فرایند ترشح و تجزیه فاکتور VIII را تقویت میکنند.
بخشی از پروتئین فاکتور VIII در شبکه اندوپلاسمی میماند و در داخل سلول تجزیه میشود. بخش دیگر، وارد دستگاه گلژی شده و تغییرات فراوانی روی آن انجام میگیرد، تا این که زنجیرههای سنگین و سبک، تولید و کربوهیدراتها، اصلاح شوند. افزوده شدن سولفات به تیروزینهای زنجیرههای سنگین و سبک، به منظور فعالیت کامل پیشانعقادی ضروری است و ناحیه سولفاته شده در فعل و انفعالات ترومبین دارای نقش است. این سولفاته شدن نواحی حاوی تیروزین پس از ترجمه، روی فعالیت پیشانعقادی فاکتور VIII و فعل و انفعالات آن با فاکتورvon Willebrand factor (vWF) تاثیر میگذارد.
فاکتور vWF
فاکتور VIII به همراه vWF به صورت کمپلکسی غیرکووالانسی در پلاسما در گردش است. vWF نقش مهمی در عملکرد، تولید، پایداری، شکل و ایمنیزایی فاکتور VIII دارد. vWF، آنتیژن مرتبط با فاکتور VIII (FVIII-related antigen (FVIII-R)) نامگذاری شده است. ترمینولوژی مربوطه برای فاکتور VIII، FVIII-coagulant (FVIII-C) میباشد.
به نظر میرسد که vWF، کنار هم قرار گرفتن زنجیرههای سبک و سنگین فاکتور VIII را افزایش داده و باعث ترشح موثرتر آن از شبکه اندوپلاسمی میشود. vWF همچنین باعث تبدیل فاکتور VIII به اجسام Weibel-Palade میگردد که محل ذخیره داخل سلولی vWF است. vWF در پلاسما، فاکتور VIII را پایدارتر کرده و از آن، در برابر تجزیه محافظت میکند. نیمهعمر فاکتور VIII در حضور پروتئین vWF نرمال، تقریبا ۱۲ ساعت است. این در حالی است که در نبود آن، نیمهعمر FVIII-C به ۲ ساعت کاهش مییابد.
آبشار انعقادی
وظیفه سیستم انعقادی، تولید لخته فیبرین پایدار در محل آسیب است. مکانیسم انعقاد دارای دو مسیر است: داخلی و خارجی. تصویر زیر را مشاهده کنید.
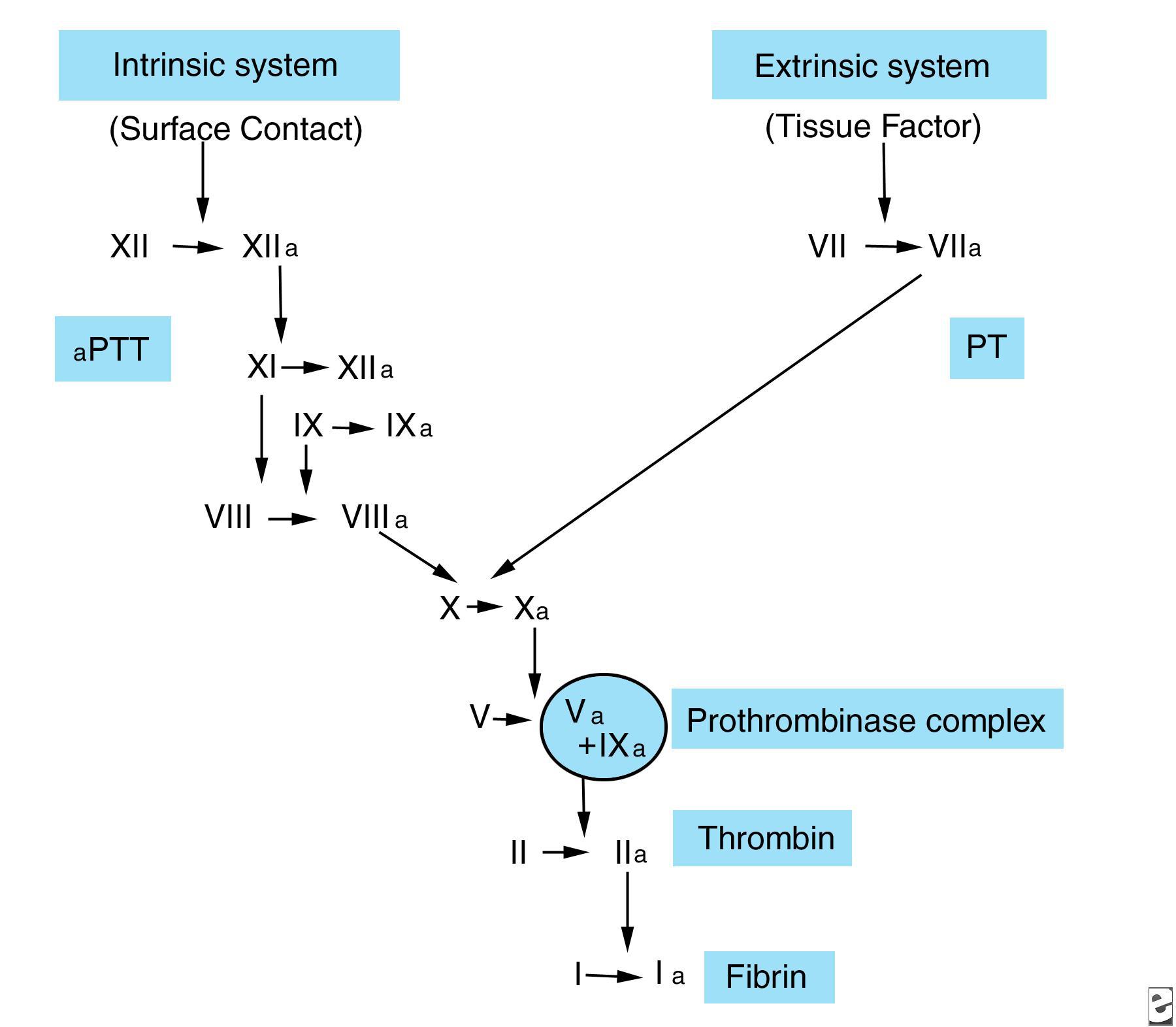
فاکتور XIIa، در ارتباط با کینینوژن با وزن مولکولی بالا (HMWK= High Molecular Weight Kininogen)، prekallikrein (PK) را به kallikrein تبدیل کرده و فاکتور XI را فعال میکند. فاکتور XI فعال نیز، طی واکنشی وابسته به کلسیم، فاکتور IX را فعال می سازد. فاکتور IXa توانایی اتصال به فسفولیپیدها را دارد؛ از این رو، فاکتور X در سطح سلول فعال میشود. فعالسازی فاکتور X نیازمند کمپلکسی از فاکتور IXa، فاکتور VIII فعالشده با ترومبین، یونهای کلسیم و فسفولیپیدها است.
در مسیر خارجی، تبدیل فاکتور X به فاکتور Xa نیازمند فاکتور بافتی (TF) و یا ترومبوپلاستین، فاکتور VII و یونهای کلسیم است. TF از سلولهای آسیبدیده رهاسازی میشود و کمپلکسی لیپوپروتئینی است که به عنوان رسپتوری در سطح سلول برای فاکتور VII عمل کرده و منجر به فعال شدن آن میگردد. TFهمچنین فاکتور X را جذب کرده و باعث افزایش واکنش میان فاکتور VIIa، فاکتور X و یونهای کلسیم میشود. قطعات فاکتورهای IXa و XII نیز میتوانند فاکتور VII را فعال نمایند.
در طی مسیر مشترک، فاکتور Xa (که از طریق مسیر داخلی و یا خارجی تولید شده است)، به همراه فسفولیپیدها، یون های کلسیم و فاکتور Va فعالشده در اثر ترومبین، تشکیل کمپلکس پروترومبیناز را میدهند. این کمپلکس، پروترومبین را به ترومبین و قطعات پروترومبین ۱ و ۲ میشکند.
ترومبین، فیبرینوژن را به فیبرین تبدیل کرده و فاکتورهای VIII، V و XIII را فعال میکند. حاصل شکستن پپتیدهای A و B توسط ترومبین، فیبرینوپپتیدهای A و B هستند که منجر به تشکیل مونومرهای فیبرین و پلیمریزه شدن آنها به صورت شبکهای از فیبرین میشوند. لخته حاصل توسط فاکتور XIIIa و اتصالات متقاطع رشتههای فیبرین مجاور، پایدار میشود.
به علت فعل و انفعالات پیچیده ای که میان مسیرهای داخلی و خارجی وجود دارد (فاکتور IXa، فاکتور VII را فعال میکند)، تصور میشود که احتمالا تنها یک مسیر داخلی in vivo وجود داشته باشد، ولی با مکانیسمهای مختلفی فعالسازی شود. تصویر زیر را مشاهده کنید.
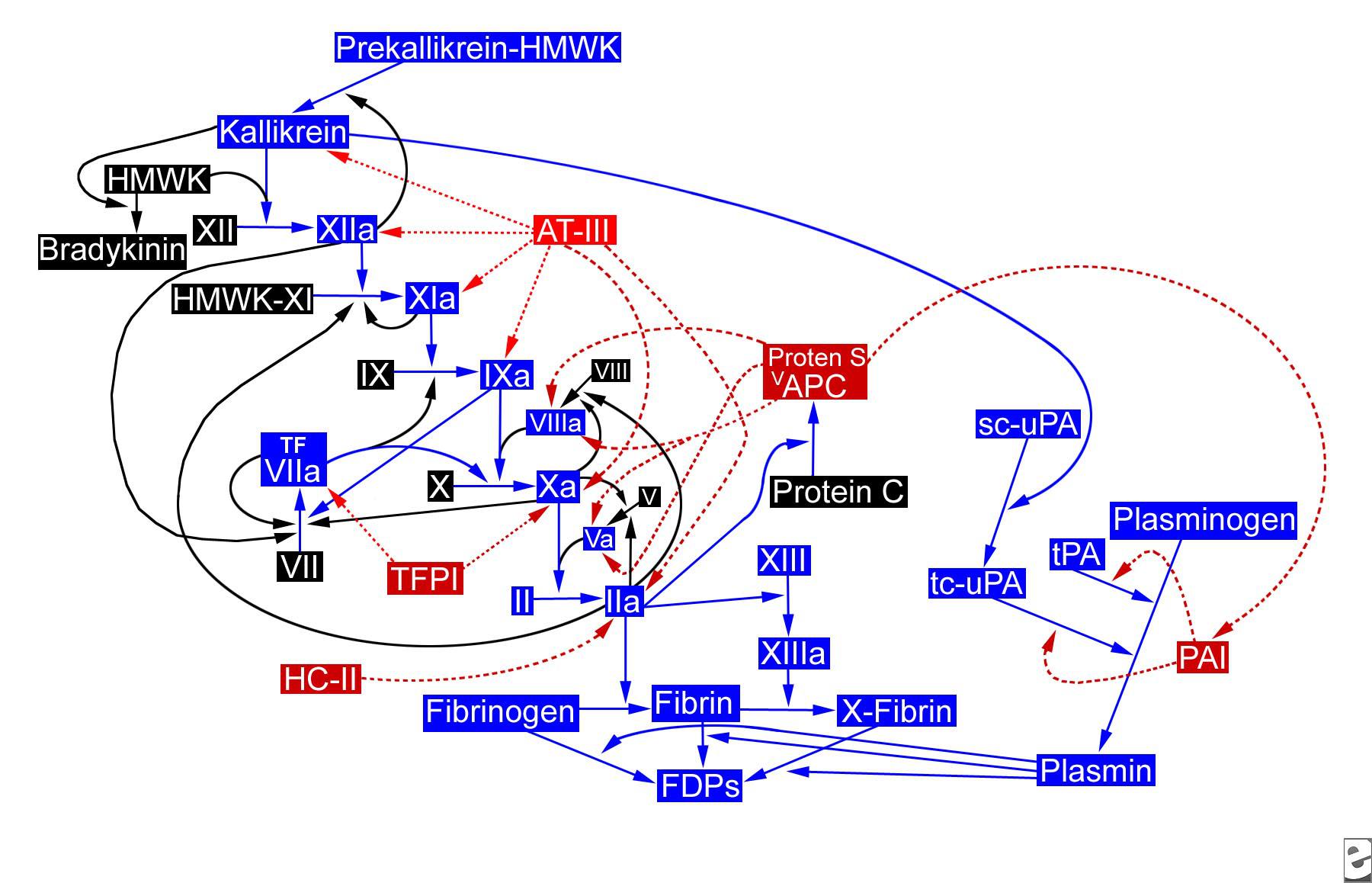
مسیر هموستاتیک. APC = activated protein C (APC)؛ AT-III = antithrombin III؛ FDP = fibrin degradation products؛ HC-II = heparin cofactor II؛ HMWK = high-molecular-weight kininogen؛ PAI = plasminogen activator inhibitor؛ sc-uPA = single-chain urokinase plasminogen activator؛ tc-uPA = two-chain urokinase plasminogen activator؛ TFPI = tissue factor pathway inhibitor؛ tPA = tissue plasminogen activator.
FVIII و فاکتور IX در فرم غیرفعال خون در گردش خون حضور دارند. در صورت فعال سازی، این دو فاکتور با کمک یکدیگر، فاکتور X را که آنزیمی کلیدی در کنترل تبدیل فیبرینوژن به فیبرین است، شکسته و فعال میکنند. در نتیجه، در نبود FVIII، تشکیل لخته به شدت با مشکل مواجه شده و متعاقبا خونریزی ایجاد میشود.
وراثت هموفیلی A
ژن FVIII که F8C نام دارد، در بازوی بلند کروموزوم X و ناحیه Xq28 قرار گرفته است. این ژن به صورتی غیرعادی بزرگ بوده و ۱۸۶ کیلوباز از کروموزوم X را دربرمی گیرد. F8C شامل ۲۶ اگزون و ۲۵ اینترون بوده و شکل نهایی FVIII دارای ۲۳۳۲ آمینواسید میباشد. تصویر زیر را مشاهده نمایید.

حدودا ۴۰ درصد از موارد کمبود شدید FVIII، ناشی از جهشهای واژگونی بزرگی هستند که ژن FVIII را مختل میکنند. جهشهای حذف و اضافه و نقطهای، 50-60 درصد سایر ناهنجاریهای F8C که منجر به هموفیلی A میشوند را تشکیل میدهند.
سطوح پایین FVIII ممکن است در نتیجه نقایصی خارج از ژن FVIII نیز ایجاد شود. به عنوان مثال، در بیماری von Willebrand نوع IIN، شاهد نقص مولکولی در دومین متصل شونده به فاکتور von Willebrand هستیم.
هموفیلی A
کمبود FVIII، وجود FVIIIهای فاقد عملکرد و یا مهارکنندههای FVIII، منجر به اختلال در فرایند طبیعی آبشار انعقادی می شود. در نتیجه، وقوع تروما باعث خونریزی بیش از حد شده و یا حتی در موارد شدید، خونریزیهای خودبهخودی روی خواهد داد. هموراژ میتواند در مفاصل (مانند زانو و آرنج)، عضلات، سیستم عصبی مرکزی و سیستمهای گوارشی، ادراری-تناسلی، ریوی و قلبی-عروقی به وجود آید. خونریزیهای داخل جمجمهای بیشتر در بیماران زیر ۱۸ سال اتفاق میافتد و میتواند مرگبار باشد.
خونریزیهای داخل مفصلی
از مشخصات اصلی هموفیلی، ایجاد خونریزی در داخل مفاصل است. این خونریزی دردناک بوده و منجر به التهاب طولانی مدت مفاصل و تخریب آنها میشود.
سلولهای سینوویال انسان مقادیر بالایی از مهار کننده مسیر فاکتور بافتی (tissue factor pathway inhibitor) را ترشح میکنند که منجر به مهار با درجه بالای فاکتور Xa میگردد. این امر، مفاصل افراد هموفیلی را مستعد خونریزی میکند. احتمالا علت بهبود سریع بیماران واجد مهارکنندههای FVIII که همارتروز حاد دارند، در پاسخ به تزریق فاکتور VIIa نیز همین موضوع باشد.
خونریزی داخل مفاصل میتواند به التهاب غشای سینوویال بینجامد، که خود این اختلال نیز خونریزیهای بیشتری را باعث خواهد شد. مفصلی که به طور مکرر خونریزی داشته است (طبق یکی از تعاریف، حداقل ۴ خونریزی در مدت شش ماه)، مفصل هدف نامیده می شود که به طور معمول زانو است.
همارتروزهای مکرر منجر به هایپرتروفی پیش رونده غشای سینوویال، رسوب هموسیدرین، فیبروز و آسیب به غضروف به همراه تولید کیست در لایه استخوانی واقع در زیر غضروف مفصلی (subchondral bone-cyst) میشود. بدشکلی دائمی مفصل، از دست رفتن حرکت آن و اندامهایی با طول نابرابر از نتایج این اختلال می باشند.
مهارکنندهها
تقریبا ۳۰ درصد از بیماران مبتلابه نوع شدید هموفیلی A، مهارکنندههایی از نوع آلوآنتی بادی را ترشح میکنند که میتوانند به FVIII متصل شوند. این مهارکنندهها معمولا از نوع IgG (خصوصا زیردسته IgG4) هستند و میتوانند اثرات انعقادی درمانهای جایگزینی فاکتور VIII را خنثی کنند. با این حال مهارکنندهها، همانند پدیدهای که در مورد کمپلکسهای ایمنی در گردش دیده میشود، کمپلمان را فعال نکرده و موجب آسیب ارگانها نمیشوند.
مهارکنندهها در سنین پایین (حدودا ۵۰ درصد موارد تا ۱۰ سالگی) و خصوصا در بیمارانی که FVIIIشان زیر یک درصد است، وجود دارند. فاکتورهای ژنتیکی و محیطی هر دو تعیینکننده شیوع تولید مهارکنندهها هستند. ناهنجاریهای مولکولی خاصی مانند حذفهای ژنی، جهش در کدون پایان و جهشهای تغییر چارچوب، با افزایش احتمال ایجاد مهارکنندهها در ارتباط میباشند. به علاوه، این احتمال در کودکان سیاهپوست بیشتر است. جهشهای بدمعنی نیز باعث افزایش اندک خطر تولید مهارکنندهها میگردند.
ارتباط استفاده از فرآوردههای خونی خاص با خطر تولید مهارکنندهها، همچنان موردبحث است. در مطالعهای که روی ۵۷۴ بیمار مبتلا به هموفیلی A شدید که ۱۷۷ نفر از آنان مهارکنندهها را تولید میکردند انجام شد، نشان داد که در صورت استفاده از هر دو فرآورده FVIII نوترکیب و مشتق از پلاسما، خطر به وجود آمدن مهارکنندهها یکسان میباشد. هیچ ارتباطی میان تولید مهارکنندهها و محتوای فاکتور von Willebrand محصولات خونی مختلف اعم از فرآوردههای نوترکیب و مشتق از پلاسما و نیز برندهای مختلف FVIII مشاهده نشده است. با این حال برخلاف انتظار، مهارکنندهها در محصولات نسل دوم نوترکیب full-length بیشتر از محصولات نسل سوم، تولید شدند.
مطالعهای که روی ۳۰۳ کودک مبتلا به هموفیلی درمان شده با محصولات نوترکیب، که پیشتر تحت درمان نبودند و یا کمترین میزان درمان روی آنها صورت گرفته بود، نشان داد که این محصولات با افزایش خطر به وجود آمدن مهارکنندهها همراه هستند. با این حال European Medicines Agency (EMA) Pharmacovigilance Risk Assessment Committee (PRAC) اظهار دارد که ارتباط شفاف و پیوستهای میان تفاوت در تولید مهار کنندهها در صورت استفاده از این دو دسته از داروهای فاکتور VIII وجود ندارد: آنهایی که از پلاسما مشتق شده اند و آنهایی که با استفاده از تکنولوژی DNA نوترکیب تولید شدهاند.
در ایالات متحده سطح مهارکنندههای FVIII غالبا توسط متد Bethesda سنجیده میشود. در این متد، یک واحد Bethesda (BU) معادل مقادیری از آنتیبادی است که نیمی از FVIII ها را در مخلوطی حاوی مقادیر یکسانی از پلاسمای نرمال و پلاسمای بیمار، در مدت زمان ۲ ساعت و دمای ۳۷ درجه سانتیگراد از بین میبرد. سطح مهارکننده ، بسته به این که کمتر و یا بیشتر از 5BU باشند، به ترتیب به صورت تیتر پایین و بالا توصیف میگردد. تیترهای بالای مهارکننده معمولا خیلی بیشتر از 5BU میباشند.
تکنیک تغییریافته Nijmegen از پلاسمای فاقد FVIII immunodepleted به جای بافر نمکی ایمیدازول استفاده میکند. به این نحو میتوان از تحت کنترل بودن pH و جلوگیری از از دست رفتن فعالیت FVIII-C به طرقی غیر از آنتی بادی موجود علیه آن در طی دوره ۲ ساعته انکوباسیون، اطمینان حاصل کرد. آزمایش Bethesda تیتر اتوآنتیبادی VIII را به علت خصوصیاتی که دارد، نسبت به آنتیبادی هموفیلیک، معمولا کمتر از حد واقعی تخمین میزند. آزمایش Oxford شکل تغییریافته دیگری از آزمایش Bethesda میباشد.
هموفیلی اکتسابی
هموفیلی اکتسابی، به تولید مهارکنندهها (اتوآنتی بادیها)ی FVIII در افرادی اطلاق میشود که سابقه کمبود FVIII را نداشته اند. این بیماری میتواند ایدیوپاتیک باشد که در این صورت در افراد بالای ۵۰ سال بیشتر رخ میدهد. هموفیلی اکتسابی میتواند با بیماری کلاژن واسکولار و یا دوره نزدیک به زایمان و نیز واکنش به داروهایی مانند پنیسیلین در ارتباط باشد. تیترهای بالای اتوآنتیبادیهای FVIII ممکن است به علت بدخیمیهای lymphoproliferative نیز رخ دهند.
اتیولوژی
هموفیلی A در اثر وقوع جهش ژنتیکی، به صورت ارثی یا اکتسابی، ایجاد شده و منجر به از دست رفتن عملکرد فاکتور VIII و یا کمبود آن میگردد؛ و یا اینکه در اثر اکتساب مهارکنندهای حاصل میشود که متصلشونده به فاکتور VIII است. در حالات ژنتیکی بیماری، حدودا بیش از یک سوم موارد در نتیجه جهشهای de novo که در کروموزوم X مادر حضور ندارند، حاصل میشوند.
مقادیر ناکافی فاکتور VIII باعث تولید ناکافی ترومبین توسط کمپلکس FIXa و FVIIIa و از طریق مسیر داخلی آبشار انعقادی می شود. این مکانیسم، در ترکیب با تاثیر ناشی از مهارکننده مسیر فاکتور بافتی، باعث وقوع اختلالی شدید در ایجاد انعقاد در پاسخ به تروما شده و در موارد شدید هموفیلی، خونریزیهای خودبهخودی رخ میدهد.
هموفیلی A الگوی وراثتی وابسته به X مغلوب دارد. ژن مربوط به FVIII در بازوی بلند کروموزوم X و در باند q28 قرار دارد. ژن فاکتور VIII از بزرگترین ژنها است و 0.1% مولکول DNA تشکیلدهنده کروموزوم X را شامل میشود؛ ۱۸۶ کیلوباز طول داشته و حاوی ناحیه کدکننده ۹ کیلوبازی که شامل ۲۶ اگزون است، میباشد. پروتئین نهایی، ۲۳۳۲ آمینواسید داشته و وزن مولکولی آن برابر ۳۰۰ کیلودالتون است. این پروتئین، دارای ۳ دومین A، یک دومین B و دو دومین C است.
اینترون ۲۲ ژن فاکتور VIII حاوی دو ژن دیگر نیز هست. ژن نخست، F8A نام دارد و در جهتی مخالف خود ژن فاکتور VIII رونویسی میشود. ژن دوم، F8B بوده و در جهت مشابه ژن فاکتور VIII رونویسی میشود. توالیهایی به نام A2 و A3، که از لحاظ توالی هومولوگ F8A میباشند، روی کروموزوم X حضور دارند و نسبت به ژن فاکتور VIII ۳۰۰ کیلوباز، تلومری میباشند.
نوترکیبی هومولوگ ژن فاکتور VIII که با واژگونی و کراساوور توالی F8A واقع در اینترون ۲۲ و توالی هومولوگ آن در سمت دیستال کروموزوم X همراه است، منجر به شکستگی ژن فاکتور VIII و قرارگیری دو قطعه آن در جهاتی مخالف هم میشود. این امر توالی کدکننده سالم فاکتور VIII را مخدوش کرده و امکان تهیه رونوشت کامل از آن را از بین میبرد. در نتیجه عملکرد پروتئین نیز از دست خواهد رفت.
جهش در اینترون ۲۲، هنگام اسپرماتوژنز ایجاد میشود و علت شایعی در کمبود شدید فاکتور VIII میباشد. این جهش تقریبا در ۴۰ درصد بیماران وجود دارد و با آنالیز DNA بیمار با استفاده از تکنیک ساترن بلاتینگ به آسانی تشخیص داده میشود. چنین بیمارانی مستعد تولید مهارکنندههای فاکتور VIII هستند.
طی یکی از مطالعات، مشخص شد که منشا تمام واژگونیهای شناسایی شده، جهشهایی است که حین اسپرماتوژنز پدربزرگ مادری رخ داده است. این موضوع تایید کننده این فرضیه است که بازوی بلند کروموزوم X جفتنشده، در مقایسه با کروموزوم X جفتشده، با احتمال بیشتری تحت تاثیر واژگونیهای داخل کروموزومی قرار میگیرد. در افراد با هموفیلی اسپورادیک شدید، که به علت واژگونی رخ داده است، اکثریت مادران ناقل این جهش میباشند.
درک منشا پدری جهشهای واژگونی میتواند در مشاورههای ژنتیکی کمک کننده باشد.
جهشهای فراوان دیگری نیز توصیف شدهاند. جهشهای نقطهای بسته به تاثیری که روی عملکرد ژن فاکتور VIII میگذارند، میتوانند منجر به کمبود خفیف، متوسط و یا شدید فاکتور VIII شوند.
جهشهای بدمعنی، مانند جایگزینیهای تکبازی G به A، ترکیب آمینواسیدی مولکول را تغییر داده و پروتئینی فاقد عملکرد را تولید مینمایند. در این حالت، آنتیژن FVIII حضور دارد اما فعالیت FVIII دچار کاهش میشود. این جهشها با کاهش خفیف، متوسط و یا شدید فاکتور VIII و ایجاد مهارکنندههای آن همراه میباشند. تجمع پروتئینهای فاکتور VIII در داخل سلول که در نتیجه جهشهای بدمعنی Arg 593→Cys و Asn 618→Ser القا میشود، همچنین منجر به کاهش cross-reacting material در موارد شدید هموفیلی A میگردد.
حذفهای ژنی به کمبود فاکتور VIII منجر میشوند و حذفهای بزرگتر نیز هموفیلی شدیدی را ایجاد میکنند که فاقد هر گونه آنتی ژن قابل تشخیص فاکتور VIII میباشد. چنین بیمارانی بیشتر از سایرین مهارکنندهها را ایجاد خواهند کردند. جهشهای اضافه در ژن فاکتور VIII شیوع چندانی ندارند، اما در صورت وقوع میتوانند منجر به هموفیلی A شدید شوند. جهشهای بیمعنی و abnormal splicing (اختلال در پیرایش) نیز امکان وقوع دارند. سایر عوامل ایجادکننده بیماری در دست بررسی میباشند.
نقص همزمان فاکتور V و فاکتور VIII (Combined factor V and factor VIII deficiency)
Combined FV and FVIII deficiency اختلال اتوزومی مفلوبی است که در هم در زنان و هم در مردان درگیر، ایجاد تظاهرات بالینی میکند. این اختلال، ناشی از وقوع جهش در یکی از این دو ژن است: lectin mannose binding protein 1 (LMAN1) و multiple coagulation factor deficiency 2 (MCFD2). این دو ژن پروتئینهایی را کد میکنند که در نقلوانتقال داخل سلولی FV و FVIII دخیل میباشند. خود فاکتورهای انعقادی در این بیماری نرمال هستند.
جمعیتهای متاثر از بیماری
هموفیلی A شایعترین اختلال ژنتیکی وابسته به X مغلوب و دومین اختلال شایع ناشی از کمبود فاکتورهای انعقادی پس از بیماری von Willebrand است. تقریبا یک نفر از هر ۵۰۰۰ نوزاد پسر، با هموفیلی A متولد میشود. تقریبا ۶۰ درصد افراد مبتلا به هموفیلی A، نوع شدید آن را تحمل میکنند. تمام نژادها و اقوام به میزان یکسانی تحت تاثیر هموفیلی قرار میگیرند.
درمانهای استاندارد
درمان
افراد مبتلا به هموفیلی A به مراکز درمان هموفیلی که federally-funded هستند، ارجاع داده میشوند. این مراکز تخصصی خدمات مراقبتی جامعی را برای افراد با هموفیلی فراهم میکنند؛ از جمله برنامههای درمانی اختصاصی، مانتیور بیماران و پیگیری (follow-up) وضعیت آنها و state-of-the-art medical care. تحت درمان گرفتن در این مراکز، بیماران و اعضای خانواده آنها را از بابت انجام مراقبت توسط تیمی حرفهای از مراقبان سلامت (شامل پزشکان، پرستاران، فیزیوتراپیستها، مددکاران اجتماعی و مشاوران ژنتیکی) که در درمان بیماران هموفیلی کارآزموده هستند، مطمئن میکند. مشاوره ژنتیک برای فرد بیمار و خانواده وی بسیار سودمند خواهد بود.
علیرغم نبود درمانی قطعی برای هموفیلی A، درمانهای فعلی بسیار موثر هستند. درمانهای صورت گرفته شامل جایگزینی پروتئین انعقادی مختلشده و پیشگیری از عوارض مرتبط با بیماری است. این پروتئین جایگزین میتواند از طریق فاکتور VIII نوترکیب که به صورت آزمایشگاهی ساخته میشود، تهیه شود. بسیاری از پزشکان و سازمانهای سلامتی مردم نهاد (voluntary health organizations) استفاده از فاکتورهای VIII نوترکیب را ترجیح میدهند. علت این موضوع، عدم وجود ترکیبات مشتق از خون انسان در این محصولات میباشد. فاکتور VIII همچنین میتواند از پلاسمای فریزشدهای که از خون اهداکنندگان به دست میآید، تهیه شود. خونهای اهدایی میتوانند انتقالدهنده ویروسهایی مانند هپاتیت باشند. البته امروزه با وجود تکنیکهای غربالگری و تیمار؟ treating خونهای اهدایی، ریسک چنین حوادثی به شدت پایین آمده است.
FDA تاکنون چندین نوع از فاکتورهای VIII نوترکیب را تایید کرده است؛ از جمله Helixate®FS (CSL Behring)؛ Recombinate® (Baxter)؛ Kogenate®FS (Bayer HealthCare)؛ Advate® (Baxter)؛ ReFacto® (Pfizer)؛ Eloctate® (Biogen-Idec) و Xyntha® (Pfizer). محصولات مشتق از پلاسمای خون انسان عبارتند از: Monarc-M (Baxter)؛ Monoclate-P® (CSL Behring)؛ Hemofil M (Baxter) و Koate-DVI (Kedrion).
Nuwiq دارویی است که به صورت داخل وریدی برای کودکان و بزرگسالان به کار میرود و در سال ۲۰۱۵ مورد تایید FDA قرار گرفته است. این درمان توسط Octapharma تولید میشود.
در سال ۲۰۱۵، Adynovate که در نوجوانان و بزرگسالانی که بالاتر از ۱۲ سال هستند کاربرد دارد، مورد تایید قرار گرفته است و توسط Baxalta U.S. Inc تولید میشود.
در سال ۲۰۱۶،FDA داروی Kovaltry Antihemophilic که فاکتوری نوترکیب در درمان هموفیلی A در کودکان و بزرگسالان است را تایید کرد.
در سال ۲۰۱۸، Jivi که یک فاکتور VIII نوترکیب است، به منظور استفاده پروفیلاکتیک، استفاده در مواقع ضروری (on demand) و نیز مدیریت خونریزی پیش از جراحی، در افراد بالای ۱۲ ساله مبتلا به هموفیلی A تایید شد. هر دو داروی Jivi و فاکتور Kovaltry Antihemophilic توسط Bayer تولید میشوند.
بیماران مبتلا به انواع خفیف و متوسط هموفیلی A در مواقع لازم و به منظور درمان خونریزیهای ایجادشده، میتوانند تحت درمان جایگزینی قرار بگیرند. این نوع از درمان، ‘on demand’ therapy نام دارد. برخی از بیماران مبتلا به نوع شدید هموفیلی A به صورت دورهای و در فواصل زمانی منظم، فاکتور VIII دریافت میکنند تا از خونریزیها و عوارض مرتبط با آنها مانند آسیب مفاصل، پیشگیری به عمل آید. این نوع از درمان، درمان پروفیلاکتیک نامیده میشود.
میتوان والدین و بیماران را به منظور انجام تزریقات در منزل آموزش داد.این موضع خصوصا در افراد با نوع شدید بیماری بسیار ضروری است؛ چون تزریق کنسانتره فاکتور VIII بیشترین تاثیر را در مدت زمان یک ساعت پس از شروع خونریزی میگذارد. در کل، درمان سریع از این جهت که درد و آسیب به مفاصل، عضلات و سایر بافتها و ارگانهای درگیر را کاهش میدهد، اهمیت دارد.
برخی از بیماران با هموفیلی A خفیف، ممکن است با desmopressin (DDAVP) تحت درمان قرار بگیرند. این دارو، سنتتیک بوده و مشتق از هورمون وازوپرسین میباشد. Desmopressin سطح فاکتور VIII در پلاسما را افزایش میدهد و ممکن است به صورت داخل وریدی و یا به صورت اسپری بینی اعمال شود. داروهایی که به antifibrinolytics معروفند، سرعت تجزیه فاکتورهای انعقادی در خون را کاهش میدهند و میتوانند در درمان افراد مبتلا هموفیلی A خفیف مورد استفاده قرار بگیرند.
مهارکنندهها
در برخی از موارد (حدودا ۳۰ درصد از افراد مبتلا به هموفیلی A شدید)، مهارکنندههایی علیه درمان جایگزین فاکتور VIII در بدن بیماران پدید میآید. مهارکنندهها از جنس آنتیبادیها هستند. آنتیبادیها پروتئینهایی اختصاصی هستند که توسط سیستم ایمنی بدن تولید میشوند و به منظور مقابله با عوامل بیگانه و مهاجم مانند باکتریها و توکسینها به کار میروند.
این احتمال وجود دارد که سیستم ایمنی فاکتور VIII جایگزین را به عنوان بیگانه تلقی کرده و این آنتیبادیها (مهارکنندهها) را علیه آن تولید کند. مهارکنندهها فاکتورهای جایگزین را هدف قرار داده و آنها را نابود میکنند.
تولید مهارکنندهها میتواند با واکنشهای آلرژیک خفیف تا شدید همراه شود. مهارکنندهها با نام آلوآنتیبادی نیز شناخته میشوند. دلایل تولید مهارکنندهها در بدن فرد بیمار، به علت دخالت فاکتورهای چدگانه در آن، پیچیده بوده و به طور کامل شناخته نشده است. خطر ایجاد مهارکنندهها در طول زندگی فرد بیمار متغیر است. به منظور تعیین دقیق مکانیسمهای توسعه مهارکنندهها در برخی از بیماران مبتلا به هموفیلی A مطالعات بیشتری موردنیاز است.
مهارکنندهها میتوانند به میزان زیادی از تاثیر درمانهای جایگزینی فاکتورهای انعقادی بکاهند. در چنین مواردی، از درمانهای دیگری به منظور کاهش خونریزی استفاده میشود و گاهی اوقات نیز اقداماتی برای حذف این آنتیبادیها صورت میگیرد (تحمل ایمنی).
سطح آنتی بادی مهارکننده در بیماران قابل سنجش است و تیتر نامیده میشود. تیتر مهارکننده با واحدی اختصاصی به نام Bethesda بیان میشود. هر چقدر تعداد واحدهای Bethesda بیشتر باشند، میزان مهارکنندهها نیز بیشتر هستند. مهارکنندهها بسته به میزان تحریک سیستم ایمنی میزبان در هر بار مواجهه با فاکتور VIII به دو دسته high-responding و low-responding تقسیمبندی میگردند. در صورتیکه پاسخ ایمنی قدرتمند باشد، سطح مهارکنندهها به میزان زیادی افزایش مییابد و پاسخ سیستم ایمنی high-responding تلقی میشود. در حالت مقابل، پاسخ سیستم ایمنی میتواند ضعیف و درنتیجه low-responding باشد.
در صورتیکه تیتر آنتیبادی بسیار پایین و زیر ۵ واحد Bethesda بوده و low-responding باشد، خونریزیهای چنین بیماری میتواند با جایگزینی فاکتور VIII در دوزهای بالا، درمان شود. در افراد با تیتر آنتیبادی بالا (بالاتر از ۵ واحد Bethesda)، جایگزینی فاکتور VIII چندان موثر نیست.
در افراد با تیتر آنتی بادی بالا، غالبا از داروهای bypassing (کنسانتره فاکتورهایی که کمبود فاکتور VIII را بایپس می کنند) برای کنترل خونریزیها استفاده میشود. داروهای bypassing موجود عبارتند از: فاکتور VII نوترکیب فعال شده (rFVIIa or NovoSeven® RT) و یا کنسانتره کمپلکس پروترومبین فعالشده (aPCC or FEIBA®) این درمانها برای همه افراد موثر واقع نشده اند.
FDA با داروی NovoSeven® RT، که نسخه نوترکیب فاکتور VII فعال است، به منظور درمان هموفیلی A موافقت کرده است. از آنجایی که این دارو به صورت آزمایشگاهی تولید میشود، حاوی پلاسما و خون انسانی نیست. در نتیجه ریسک انتقال ویروسهای منتقلشونده از راه خون و سایر پاتوژن ها کاهش مییابد. NovoSeven به خوبی تحمل میشود و عوارض جانبی کمی دارد. خطر اثرات جانبی ترومبوتیک (تشکیل ترومبوز) در بیماران هموفیلی درمان شده با این دارو، زیر یک درصد است. NovoSeven® RT توسط کمپانی دارویی Novo Nordisk تولید میگردد.
aPCC کمپلکس ضدمهارکننده مشتق از پلاسمایی است که حاوی انواعی از فاکتورهای انعقادی فعال میباشد. این فاکتورها باعث میشوند که دارو تعدادی از مراحل تشکیل لخته، از جمله مرحلهای که نیازمند فاکتور VIII است را بای پس کند. aPCC به نحوی پردازش میشود که ویروسها و پاتوژنهای مشابه موجود در آن غیرفعال شوند و تشکیل ترومبوز در صورت استفاده از آن به ندرت رخ میدهد. تنها aPCC ای که در حال حاضر در ایالات متحده موجود میباشد، FEIBA® که توسط Baxter Healthcare Corporation تولید میشود.
در سال ۲۰۱۷، با استفاده از Hemlibra (emicizumab-kxwh) به منظور جلوگیری از خونریزی و یا کاهش دفعات آن در بزرگسالان و کودکانی که مبتلا به هموفیلی A بوده و مهارکنندهها را تولید میکردند، موافقت شد. تولید این دارو بر عهده Genentech, Inc است.
در برخی از بیماران که مهارکنندهها را تولید میکنند، از درمانی به نام القای تحمل ایمنی استفاده میشود. این درمان به منظور حذف مهارکنندهها استفاده میشود و امکان استفاده از درمانهای جایگزینی فاکتور VIII را به بیمار میدهد. طی فرایند القای تحمل ایمنی، بیمار طی مدت زمانی که ممکن است از چندین ماه تا چندین سال طول بکشد، در معرض دوز بالایی از فاکتور VIII قرار میگیرد. هدف از انجام این فرایند، تعلیم سیستم ایمنی است؛ به نحوی که بتواند درمان جایگزینی با فاکتور VIII را بدون تولید مهارکنندهها بپذیرد. کاستیهای القای تحمل ایمنی، هزینه بالا ، درد و رنج بیمار و زمانبر بودن آن است. با این حال، القای تحمل ایمنی در حذف مهارکنندهها در حدود ۷۰ درصد موارد موثر بوده است.