کلیات بیماری
در سال 1930، ولف، پارکینسون و وایت اطلاعات یک دسته از بیماران جوان را توصیف کردند که دارای بلوک شعاعی شاخه ای بر یافته های الکتروکاردیوگرافی (ECG)، یک فاصله کوتاه PR، و پاروکسیسم تاکی کاردی بود. گزارش های موردی در مقالهها در اواخر دهه 1930 و اوایل دهه 1940 شروع شد و اصطلاح “سندرم ولف پارکینسون وایت” (WPW) در سال 1940 ساخته شد.
اگرچه “پیشزمینه” اولین بار توسط Ohnell در یک نشریه برجسته در سال 1944 ساخته شد، اصطلاح که توسط Durrer و همکاران در سال 1970 تعریف شده است، توصیفی بهتر از آنچه در مسیر تکمیلی وجود دارد (قبل از ظهور مطالعات الکتروفیزیولوژیک invusive و ablation ارائه یک درک روشن تر : “پیشزمینه وجود دارد، اگر در ارتباط با وقایع دهلیزی، تمام یا بخشی از عضله بطنی زودتر اززمان مورد انتظار توسط ایمپالس ناشی از دهلیز تحریک میشود.”
سندرم WPW در حال حاضر به عنوان یک اختلال مادرزادی که شامل وجود بافت قلبی هدایت غیر طبیعی بین دهلیز و بطن در ارتباط با تاکیکارد سوپراوانتراکالی (SVT) است، تعریف می شود. این شامل پیشزمینه است که به دلیل رسیدن یک ایمپالس دهلیز به وسیله یک سیستم هدایت طبیعی انجام می شود، اما از طریق یک اتصال عضلانی اتریوواستریک (AV)، یک مسیر جانبی (AP) نامیده می شود که از گره AV دور می شود.
یافته های کلاسیک ECG که با سندرم WPW مرتبط هستند عبارتند از:
- وجود یک فاصله کوتاه PR (کمتر از 120 میلیثانیه)
- موج QRS گسترده ای که بیش از 120 میلی ثانیه طول می کشد با شروع ناگهانی شکل موج QRS، به نام موج دلتا، در اوایل بخش QRS
- تغییرات موج ثانویه ST-T (مطابق شکل زیر)
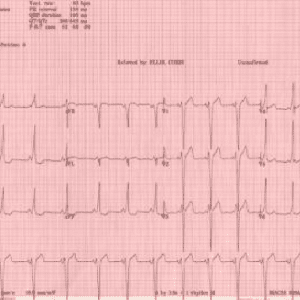
الکتروکاردیوگرام کلاسیک Wolff-Parkinson-White با PR کوتاه، QRS> 120 ms و موج دلتا.
بیماران مبتلا به سندرم WPW بالقوه در معرض خطر آریتمی های بطنی به علت هدایت در مسیر عبور جانبی قرار دارند و درنتیجه دپلاریزاسیون بطن بسیار سریع بوده، در صورتی که یک فریت سرریز دهلیزی یا فیبریلاسیون دهلیزی (AF) ایجاد شود.
برخی از بیماران دارای یک مسیر عبور مخفی هستند. اگرچه آنها دارای یک اتصال AV جانبی هستند، اما دارای یک خطای خاص نیست؛ بر این اساس، این بیماران EKG اختلالات کلاسیک سطح را ندارند. به طور معمول، این مطالعه توسط الکتروفیزیولوژیک انجام شده برای ارزیابی یا درمان SVT انجام شده است.
تنها درصد کمی از بیماران مبتلا به سندرم WPW (کمتر از 1٪) در معرض خطر مرگ ناگهانی قلب (SCD) هستند. در بیماران مبتلا به AF پیشآگهی، مطالعات الکتروفیزیولوژیک قلب و تخلیه کاتتر رادیویی (RF) ممکن است درمان شوند. ارائه های دیگر شامل SVT علامتی است که می تواند توسط تخلیه کاتتر نیز درمان شود. بیماران بدون علامت به مشاهده دوره ای نیاز دارند. شروع تخریب کاتتر پروفیلاکتیک نیز ممکن است با بروز آریتمی قلبی و احتمالا احتمال مرگ ناگهانی مرگ همراه باشد.
پاتوفیزیولوژی
مسیرهای جانبی یا اتصالات بین دهلیز و بطن چپ به علت ایجاد انسداد جنین از بافت های قلب از طریق بریدن بافت های فیبرینی است که دو اتاق را جدا می کنند. این اجازه می دهد تا انتقال الکتریسیته بین دهلیز و بطن ها در نقاطی غیر گره AV برقرار شود. عبور از طریق AP باعث می شود که تاخیر هدایت معمول بین دهلیز و بطن، که معمولا در گره AV اتفاق می افتد، دور از انتظار می رود، و بیمار را برای مبتلا به تاکی دیسترمی مستعد میسازد.
اگرچه در پیشزمینه ممکن است راه های مختلفی وجود داشته باشد، از جمله atriofascicular, fasciculoventricular, nodofascicular , یا nodoventricular که معمولا مورد استفاده قرار می گیرد بایپس مسیر راه دسترسی AV است، در غیر این صورت به عنوان شاخه کنت شناخته شده است. این یک ناهنجاری در سندرم WPW دیده می شود. یکی از ویژگی های اصلی که سندرم WPW را از سایر تاکیکاردیهای مربوط به AP متفاوت میکند، توانایی AP برای ایجاد ارتباط بین دهلیز و بطن به صورت دوطرفه است.
وجود AP یک پتانسیل برای تداخل تکرار تاکیکاردی را ایجاد می کند یا برای تاکیکاردیهای پیش زمینه در تنظیم فیبریلاسیون دهلیزی، یا SVT با یک مسیر جانبی افزودنی ایجاد می کند. این مکانیزم بازآفرینی عامل اصلی SVT است که بیماران مبتلا به پیش زمینه در معرض خطر هستند. پیدایش SVT که در حال بازگشت است، شامل وجود مسیرهای هدایت دو طرفه بین دهلیز و بطن است :
- گره AV طبیعی در مسیر
- یک یا بیش از یک مسیر جانبی AV
این مسیرها معمولا ویژگیهای رسانایی مختلف و دوره های مقاوم را نشان می دهند که باعث تسکین مجدد می شوند. دوره رفع موثر از بخش کمکی اغلب طولانی تر از مسیر هیس-پورکنژ معمولی گره AV است و قبل از اینکه مجددا راه اندازی شود نیاز به زمان دارد. درجه قبل از ظهور بر روی یک ECG سطح در یک فرد با الگو WPW می تواند با عرض QRS و طول فاصله PR محاسبه شو QRS گسترده تر یا بیشتر که با فاصله کوتاه PR با اجزای ایزوالکتریک وجود ندارد یا تقریبا غایب است، نشان می دهد که بیشتر (یا همه) دپلاریزاسیون بطن چپ از طریق قرار دادن AP به جای گره AV سیستم پورکنژ او آغاز می شود. این ممکن است با مسیرهای سمت راست دیوار معمولی که در آن قرارگیری مجرای دهلیزی نزدیک به گره sinoatrial) SA) باشد، معمول است.
با این حال، عرض QRS ممکن است متفاوت باشد، در حالی که ضربان قلب سریعترافزایش می یابد. این امکان وجود دارد که کاتلول آمینها اجازه می دهند که گره AV به افزایش (یا به طور کامل) دپلاریزاسیون بطنی با افزایش گره AV کمک کند؛ گره AV به سیستم کلی و معمولی هیس-پورکنژ متصل می شود، که در نتیجه موج QRS باریک است.
انواع SVT شامل تاکیکارد ارتودرومی (پایین سیستم هیس-پورکنژ گره AV و هدایت رتروگراد AP)، تاکیکارد ارتودرومی با پنهان AP و تاکیکاردی ضددرد (پایین تر از AP و هدایت رتروگراد به سیستم هیس-پورکنژ و گره AV در بیماران مبتلا به WPW که AP در ریه های بیمار شرکت می کنند، 95٪ SVT به علت تاکیکارد اورتودرمی است و 5٪ به علت تاکیکارد ضددردی است.
تاکیکارد Orthodromic
هنگامی که یک ایمپالس دهلیزی زودرس نارسایی به سمت بطن پیش میرود، میتواند در AP متزلزل شود، اما راه AVN / His Purkinje را نادیده میگیرد. این ضربه پس از آن AP را در حالت بازگشت قرار می دهد تا جنبش سیرک این تحریک را تثبیت کند. چنین تاکیکاردی بازگشتی اورتودرمیک توصیف می شود. انقباضات بطنی زودرس (PVCs) همچنین می تواند تاکیکاردی ارتودرومی را آغاز کند.
در تاکیکاردی ارتودرومی، مسیر طبیعی برای دپلاریزاسیون بطنی استفاده می شود و AP برای هدایت برگشتی ضروری برای بازگشت مجدد استفاده می شود. در یافته های ECG، موج دلتا وجود ندارد، مجموعه QRS طبیعی است، و امواج P به طور معمول در منحنی های پایین تر و جانبی تغییر می کنند.
تاکیکارد Orthodromic با مسیر جانبی جانبی پنهان
بعضی از AP ها قادر به انجام مداخله ای نیستند. اینها APs پنهان نامیده می شوند، چرا که “پیش بینی ظاهری” یک موج دلتا است که بر روی یک ECG 12-lead سطح ظاهر می شود. (از لحاظ فنی، مسیرهای پنهان نباید به عنوان یک سندرم WPW طبقه بندی شوند، زیرا موج دلتا وجود ندارد.) آنها حدود 30 درصد از همه SVT های ناشی از EPS را تشکیل می دهند.
اگر چه در طی ریتم سینوسی هیچ شواهدی از مسیر وجود ندارد (به عنوان مثال، هیچ پیشآگهی در ECG)، ممکن است تاکی کاردی های ارتودرومی رخ دهد. تاکیکاردی Orthodromic همچنین ممکن است در زمانی که دو یا چند اتصالات جانبی وجود دارد، و در این حالت، هدایت برگشتی ممکن است از طریق گره AV، از طریق یکی از اتصالات جانبی یا از طریق هر دو رخ دهد.
این نوع از SVT ممکن است دشوار باشد تا از یک تاکی کاردی بازتوانی تومور (AV (AVNRT، در یک ECG سطح استاندارد تشخیص دهد. در بزرگسالان، اگر ضربان قلب بالاتر از 200 و یا موج P است در بخش (ST (Tachycardia long-R-P قابل مشاهده است، ممکن است یک تاکیکاردی بازتوزیع کننده OR ) (ORTپنهان شده با AP باشد که تشخیص داده شود. با این حال، این تعیین دقیق تر با مطالعات الکتروفیزیولوژیک (EPS) ساخته شده است، و یا اگر SVT با تک PVC تکمیل شود. سایر عوامل تمایز عبارتند از:
- AVNRT قدامي: وجود موج شبه r در V1 سرب يا موج شبه S در هدایت II، III و aVF
- AVNRT خلفی: وجود اختلاف بیش از 20 میلی ثانیه در فاصله R-P بین منشاء I و III
- AVNRT وجود یک بلوک AV یا اختلال AV (غیر معمول و کوتاه مدت)، عدم حضور AP؛ بلوک شاخه ای از بسته با یک پروتز دهلیزی بدون مجرای (AA) و یا اواسط (HH)
- AVRT Orthodromic: توسعه یک بلوک شاخه بسته در حضور یک تغییر قابل توجه در فاصله زمانی ventriculoatrial (VA) (تشخیصی)، با محلی سازی AP به همان طرف بلوک.
تاکیکاردی ضددرد
شایعتر، یک دوره مقاومتر کوتاهتر در AP ممکن است باعث انسداد یک ایمپالس ناحیه اکستروپیک در مسیر طبیعی شود، با انجام آن تفاوتی از AP و سپس بازگرداندن مجدد مسیر مسیر عادیAV. این نوع تاکی کاردی به نام تاکیکاردی ضددردی نامیده می شود.
در الکتروکاردیوگرام QRS گسترده است، که منعکس کننده غلظت موج دلتا در طی ریتم سینوسی به عنوان مثال، Tachycardia گسترده QRS است. چنین تارکاردی ها دشوار است که از تاکی کاردی های بطنی جدا شوند و اغلب دارای سکته مغزی خفیف R با طول QRS بیش از 160 میلی ثانیه است.
فقط حدود 5٪ تاکیکاردی در بیماران مبتلا به سندرم WPW، تاکیکاردی ضددرد هستند؛ 95٪ باقیمانده ارتودواری هستند. حتی زمانی که AP به تنهایی در حالت عقب مانده انجام می شود، هنوز هم می تواند در مدار بازآموزی شرکت کند و با یک ریز ساختار QRS باریک یک ورید تولید کند. وجود یک تاکیکاردی ضددردی باید جستجوی دقیق یک وسیله جانبی را طی کند. حدود 10 تا 15 درصد از بیماران مبتلا به WPW دارای مسیر دوم هستند.
اتیولوژی
AP ها پدیده های مادرزادی محسوب می شوند که مربوط به شکست انعطاف پذیری بافت درون حلقه AV می شود، حتی اگر در سال های بعد از آن، تظاهرات آنها اغلب تشخیص داده می شود و به نظر می رسد آنها “به دست آوردن” هستند. در موارد نادر، سندرم WPW به دست آمده در بیماران تحت عمل جراحی مادرزادی قلبی رخ داده است، که ممکن است با توجه به اتصال AV-اپیکاردی عملکردی حاصل شود.
مطالعات خانوادگی و همچنین تحقیقات ژنتیکی مولکولی نشان می دهد که سندرم WPW همراه با اختلالات پیش رونده همراه ممکن است یک عنصر ژنتیکی داشته باشد. ممکن است به عنوان یک ویژگی خانوادگی با یا بدون نقص مادرزاد قلب (CHDs) به ارث برده شود؛ 3.4٪ از کسانی که مبتلا به سندرم WPW هستند، بستگان درجه اول با پیش پیش بینی شده هستند.
شکل خانوادگی معمولا به عنوان ویژگی غالب اتوزوم مغزی ماندلیایی به ارث می رسد. اگر چه نادر، ارثی میتوکندری نیز توصیف شده است. این سندروم همچنین ممکن است با سایر اختلالات قلبی و غیر قلبی، مانند نقص پروتز دهلیزی خانوادگی، فلج دوره ای فیزیوتراپی خانوادگی و اسکلروز توبروز به ارث برده شود.
متخصصین بالینی معتقدند که ارتباط سندرم WPW با کاردیومیوپاتی هیپرتروفی فامیلی اتوزومالی غالب وجود دارد. با این حال، تنها به تازگی یک ماده ی ژنتیکی بود که باعث پیوند کراتومیوپاتی هیپرتروفیک با سندرم WPW و مایوپاتی اسکلتی شد.
بیماران مبتلا به جهش در واحد گاما 2 واحد پروتئین کیناز فعال شده با آدنوزین مونوفسفره (AMP)، پروتئین کیناز (PRKAG2) فعال شده، کریومیوپاتی را که به واسطه هیپرتروفی بطنی، سندرم WPW، بلوک AV و بیماری سیستم هدایت سیستم دژنراتیو پیشرفته شناخته می شود، ایجاد می کند. به نظر می رسد که جهش باعث ایجاد اختلال فیبروز در حلقه می شود با انقباض گلیکوژن درون میوسیت ها، که سبب ایجاد زودرس می شود. به نظر میرسد این مورد در بیماری Pompe، بیماری Danon و سایر بیماری های ذخیره سازی گلیکوژن است.
بیماری Pompe نوزادان یا بیماری ذخیره سازی گلیکوژن نوع II یک اختلال عضلانی کشنده است که ناشی از کمبود اسید آلفا گلوکوزیداز (GAA) است. این بیماران دارای یک فاصله کوتاه PR، ولتاژ بزرگ بطن چپ (LV) و افزایش پراکندگی (QT (QTd هستند.
اخیرا، به نظر میرسد محققان یک مکان جدید را در یک خانواده با WPW، MYH6 p.E1885K شناسایی کرده اند. همه اعضای خانواده با WPW اما هیچ یک از بستگان نامشخص این نوع را نشان ندادند. انواع MYH6 با نقص پروتز دهليزي، كارديوميوپاتي و سندرم دائمي سينوس ارتباط دارد.
به نظر می رسد جهش در پروتئین غشاء 2 مرتبط با لیزوزوم (LAMP2) که سبب انباشت گلیکوژن قلب می شود، علت تعداد قابل توجهی از کاردیومیوپاتی هیپرتروفی در کودکان است، به ویژه هنگامی که میوپاتی اسکلتی، سندرم WPW یا هر دو وجود دارد.
به عنوان مثال، بیماری Danon یک میوپاتی قلب و عروق لیزوزومی مرتبط با X است؛ مردان بیشتر و بیشتر از زنان متاثر می شوند. این ناشی از جهش در LAMP2 است که باعث ضعف عضلانی پروگزیمال و آتروفی خفیف، هیپرتروفی بطن چپ، سندرم WPW و عقب ماندگی ذهنی می شود.
بیماران مبتلا به آنومالی Ebstein ممکن است سندرم WPW را ایجاد کنند. آنها اغلب دارنده موارد جانبی جانبی متعددی هستند، بیشتر در سمت راست، در قسمت پشتی سپتوم یا دیواره بعد از بطن راست بطن راست. تك كارديو عروق ارتودومايي در اين بيماران اغلب بلوك راست شاخه اي (RBBB) و فواصل طولاني مدت (ventriculoatrial) را نشان مي دهد.
در صورت وجود برخی از انواع تغییرات Bjork از روش فونت، جراحی ممکن است به صورت جراحی ایجاد شود، در صورتی که بافت دهلیزی به داخل بافت بطن چسبیده و بافت آن باقی می ماند. برخی از تومورهای حلقه AV، مانند رابدومیوم، همچنین ممکن است باعث پیشگیری شوند.
اپیدمیولوژی
آمار ایالات متحده
شيوع پيش زمینهای بطني، به نظر مي رسد که 0.1-0.3٪ باشد، يا 1-3 در هر 1000 نفر در جمعيت عمومي. برآورد بروز آریتمی در بیماران مبتلا به پیش هضم متفاوت است، در بین چندین نظرسنجی از 12 تا 80 درصد متغیر است.
میزان بروز پیش آگهی و سندرم WPW بین 0.1 تا 3 مورد در هر 1000 نفر (متوسط، 1.5 مورد در هر 1000 نفر) در افراد سالم است. این شامل تنها بیماران مبتلا به پیش داوری آشکار(موج دلتا در ECG رشته 12) است. حدود 60-70٪ از این افراد شواهدی دیگر از بیماری قلبی ندارند. تقریبا چهار مورد جدید تشخیصی سندرم WPW در هر 100،000 نفر سالانه رخ می دهد.
در بررسی ECG یافته ها از 22،500 پرسنل هواپیما سالم، 0.25٪ یافته های خود را مطابق با الگوی WPW نشان می دهد، با 1.8٪ گزارش شده بروز تکی کاردی.
موقعیت مسیرهای جانبی (AP)، در فرآیند نزولی فرکانس، (1) 53٪، دیوار چپ آزاد، (2) 36٪، posteroseptal، (3) 8٪، دیواره سمت راست، و (4) 3٪، anteroseptal.
حضور AP مخفی برای حدود 30٪ از بیماران با SVT ظاهری که برای مطالعات الکتروفیزیولوژیک (EPS) ارجاع داده شده است. این بیماران سندرم WPW “کلاسیک” ندارند، زیرا هیچ موج دلتا وجود ندارد، اما آنها دارای پتانسیل تاکیکاردی ارتودرومی هستند.
تقریبا 80٪ بیماران مبتلا به سندرم WPW دارای تاکی کاردی متقاطع هستند، 30- 15٪ فیبریلاسیون دهلیزی را ایجاد می کنند و 5٪ فتوتراپی دهلیزی دارند. VT غیر معمول است بیماران مبتلا به پرولاپس دریچه میترال دارای ارتباط با WPW هستند، اما این مکانیزم نامشخص است.
آمار بین المللی
در سراسر جهان، شیوع و شیوع سندرم WPW موازاتی است که در ایالات متحده دیده می شود.
آمار جمعیتی مرتبط با سن
سندرم WPW در افراد مختلفی یافت می شود. اکثر بیماران مبتلا به سندرم WPW در دوران شیردهی حضور دارند. با این حال، قله دوم ارائه در کودکان مدرسه و نوجوانان ذکر شده است. این توزیع سن بیستم جالب به علت از دست دادن دائمی یا گذرا از قبل از زایمان در دوران پس از زایمان در بعضی از بیماران و در دوران نوجوانی در دیگران است.
شیوع سندرم WPW با افزایش سن در نتیجه کاهش آشکار سرعت هدایت در AP کاهش می یابد. حدود یک چهارم بیماران از یک دوره 10 ساله، قبل از زایمان از دست می دهند، احتمالا به دلیل تغییرات فیبروتیک در محل تزریق بای پس عطاری با از دست دادن خواص الکتریکی هدایت الکتریکی بین اتاق های قلب. مواردی که در آن شواهد ECG قبل از زایمان به طور کامل از بین رفته است، شرح داده شده است. یک دهم بیماران مبتلا به آپنه پنهان، بیش از 10 سال در معرض ابتلا به سرطان قرار دارند.
در بیماران بدون علامت، هدایت ان تفاوتی در سراسر AP ممکن است به طور خود به خود با سن پیشرفت ناپدید می شوند (یک چهارم از بیماران از گذشت زمان انفجار بایپس بیش از 10 سال).
در بیماران با یافته های ECG غیر طبیعی که نشان دهنده سندرم WPW است، فراوانی پاروکسیسم SVT از 10٪ در افراد 20 تا 39 سال به 36٪ در افراد بالای 60 سال افزایش می یابد. به طور کلی، حدود 50٪ از بیماران مبتلا به WPW تاکیراتیتی ایجاد می کنند.
آمار جمعیتی مرتبط با جنسیت
به نظر می رسد الگو WPW بر دو جنس به طور مساوی تأثیر می گذارد؛ با این حال، سندرم WPW در مردان بیشتر دیده می شود. یک مطالعه مستند نسبت زن و مرد تقریبا 2:1. یکی دیگر از موارد گزارش شده 1.4 مورد سندرم WPW در 1000 مرد و 0.9 مورد در هر 1000 بود. در یک مطالعه سوم، شیوع سندرم WPW در مردان 3.5 برابر بیشتر بود.
پیش بینی
پس از شناسایی و درمان مناسب، سندرم WPW با پیش آگهی عالی همراه است، از جمله پتانسیل برای درمان دائمی از طریق تخلیه کاتتر رادیویی (RF).
بیماران بدون علامت که فقط قبل از زایمان در ECG هستند، پیش آگهی بسیار خوبی دارند. بسیاری از آریتمی های علامتی را در طی زمان ایجاد می کنند که می تواند با تخریب کاتتر RF و RF EPS پیشگیری شود. [16] بیماران مبتلا به سابقه خانوادگی مرگ ناگهانی قلب (SCD) یا نشانه های قابل توجهی از تاکی ریتمی یا توقف قلب، پیش بینی های بدتری دارند. با این حال، هنگامی که درمان قطعی انجام می شود، از جمله تخریب درمانی، پیش آگهی یک بار دیگر بسیار عالی است.
طبقه بندی بیخطر (مانند نظارت Holter، تست ورزش استرس) می تواند مفید باشد، اگر زودتر و کامل از دست دادن قبل از زایمان با ورزش یا تزریق پروکاریایید رخ می دهد. با این حال، این یک پیش بینی مطلق برای فقدان قسمت های آریتمی نیست.
مرگ و میر / بیماری
مرگ و میر در سندرم WPW نادر است و مربوط به SCD است. شیوع SCD در سندرم WPW تقریبا 1 در 100 مورد علائمی است که بعد از آن تا 15 سال ادامه دارد. اگر چه نسبتا غیر معمول، SCD ممکن است اولین ارائه در 4.5٪ موارد باشد.
حتی در بیماران مبتلا به WPW بدون علامت، خطر SCD بالاتر از جمعیت عمومی است. درمان پزشکی با عوامل مانند دیگوکسین ممکن است این خطر را افزایش دهد خطر ابتلا به بیماران بدون علامت پایین است و می تواند با تخلیه کاتتر پیشگیرانه از مسیر اضافی (EPS و RF تخلیه) کاهش یابد.
عوامل دیگری که به نظر میرسد بر روی خطر SCD تاثیر میگذارند عبارتند از: مجراهای متعدد بای پس، دورههای کوتاه مدت ، AF و فلوتر سرگیجه یا سابقه خانوادگی مرگ ناگهانی ناگهانی. SCD بدون علائم قبلی غیر معمول است.
علت SCD در سندرم WPW انتقال سریع AF به بطن ها از طریق AP است که موجب فیبریلاسیون بطنی (VF) می شود. AF در یک پنجم تا یک سوم بیماران مبتلا به سندرم WPW توسعه می یابد؛ دلایل این و تأثیر AP بر روی توسعه آن مشخص نیست.
با این حال، یک مطالعه فرض شده است که دو مکانیسم در پاتوژنز AF در بیماران مبتلا به سندرم WPW دخالت دارند: یکی مربوط به AP است که predispose به فیبریلاسیون پیشانی، و دیگری مستقل از AP و مربوط به افزایش آسیب پذیری دهلیزی در این افراد.
بدیهی است که AF ممکن است در بعضی از بیماران پس از تخریب موفقیتآمیز مجرای جانبی باقی بماند و در بعضی از بیماران دارای علائم باشد اما AF پس از آن خطر مشابه SCD را در معرض خطر قرار نمی دهد.
در مطالعه ای که سابقه طبیعی WPW در طولانی مدت (متوسط 6.9 سال) را در بیماران بالغی که تحت درمان با(n=872) و بدون تخلیه کاتتر (n = 1461) در مقایسه با گروه شاهد (171،179) ارزیابی شد، هم چنین در بیماران مبتلا به WPW در مقایسع با گروه شاهد میزان مرگ و میر کم، اما خطر ابتلا به AF بالا بود. خطر مرگ و میر طولانیمدت در افرادی بود که در مقایسه با گروه تحت درمان با تخلیه قرار نگرفتند، در حالی که خطر عارضه AF در گروه تخلیه بالاتر بود. بنابراین تخلیه خطر ابتلا به AF را کاهش نمی دهد.
مطابق مقالات، عوامل خطرساز توسعه AF در تنظیم سندرم WPW عبارتند از: افزایش سن (دو سن پیک برای وقوع AF تشخیص داده می شود، یکی در 30 سال و دیگری در 50 سال)، جنسیت مرد و سابقه قبلی سنکوپ
بعضی عوامل احتمال VF را افزایش می دهند، از جمله هدایت سريع AP ها و مسیرهای متعدد. مواردی نیز در ارتباط با مطالعات مری، دیگوکسین و وراپامیل گزارش شده است. چندین گزارش سندرم خودبهخود VF را در سندرم WPW مستند می کند و SVT ممکن است منجر به AF و بنابراین منجر به VF می شود ؛ با این حال، هر دو سناریو در بیمار کودکان بسیار نادر است.
احتمال ابتلا به سرطان سیناپس سریع یا آریتمی هموپکوپیک مرتبط است. حتی در صورت عدم وجود سنکوپ، قسمتهای آریتمی ممکن است بسیار علائم باشد. در اکثر بیماران، SVT به خوبی تحمل می شود و تهدید کننده زندگی نیست. با این حال، پتانسیل انسنکوپیک، ریتم مصونیت همودینامیک یا مرگ ناگهانی ممکن است بیماران مبتلا به سندرم WPW را از مشارکت در ورزش های رقابتی یا مشاغل خطرناک جلوگیری کنند تا زمانی که بستر به طور قطعی درمان شود و با روش تخلیه کاتتر درمان شود.
عوارض جانبی
عوارض عبارتند از:
- تاجیاریتمیا
- پالپیتاسیون
- سرگیجه یا انسداد
- مرگ ناگهانی قلب
- عوارض درمان دارویی (به عنوان مثال، پرواریلیت، سمیت ارگان)
- عوارض ناشی از روش های تهاجمی و جراحی
- عود
وراثت
اغلب موارد سندرم Wolff-Parkinson-White در افرادی که سابقه خانوادگی آشکار بیماری ندارند رخ می دهد. این موارد به صورت پراکنده توصیف می شوند و به ارث نمی برند.
سندرم خانوادگی Wolff-Parkinson-White فقط درصد کمی از همه موارد این وضعیت را تشکیل می دهد. شکل خانوادگی این اختلال معمولا یک الگوی غالب اتوزومی دارد که به این معنی است که یک نسخه از ژن تغییر یافته در هر سلول برای ایجاد بیماری کافی است. در اغلب موارد، فرد مبتلا به سندرم Wolff-Parkinson-White خانوادگی این بیماری را از والدین آسیب دیده به ارث برده است.
درمان های استاندارد
درمان
درمان با سندرم ولف-پارکینسون-وایت ممکن است شامل مشاهده بدون مداخله خاص، استفاده از داروهای مختلف و یک روش جراحی شناخته شده به عنوان کاتتر (رادیوفرنسی) تخلیه شود.
روش های مختلف درمانی و مداخلات ممکن است بسته به عوامل متعددی مانند نوع آریتمی موجود باشد؛ فرکانس؛ نوع و شدت علائم مرتبط؛ خطر ابتلا به قلب سن فرد و سلامت عمومی؛ و یا عناصر دیگر. تصمیمات مربوط به استفاده از مداخلات خاص باید توسط پزشکان و سایر اعضای تیم مراقبت های بهداشتی در مشاوره دقیق با بیمار، بر اساس خاصی از پرونده وی انجام شود؛ یک بحث کامل درباره مزایا و خطرات بالقوه؛ ترجیح بیمار؛ و دیگر عوامل مناسب.
در برخی از افراد بدون علائم (موارد بدون علامت) هیچ درمانی لازم نیست. برای نظارت بر عملکرد قلب ضروریات منظم، پیگیری ضروری است.
داروهای مختلفی برای کنترل قسمت های آریتمی در بعضی افراد مبتلا به سندرم WPW استفاده می شود. داروهای مشابه آنتی آریتمیک شامل آدنوزین، پروکاریامین، سوتالول، فلکایینید، ایبوتیید و آمیودارون هستند. مسدود کننده های کانال کلسیم مانند وراپامیل نیز ممکن است استفاده شود. بعضی از داروها مانند وراپامیل ممکن است خطر فیبریلاسیون بطنی را افزایش دهند و باید با احتیاط استفاده شوند.
داروهای قلبی، دیگوکسین، در بزرگسالان مبتلا به سندرم WPW مخالف است. با این حال، گاهی اوقات برای پیشگیری از درمان نوزادان مبتلا به سندرم WPW که پیش از بارداری بطنی ندارند، استفاده می شود.
در برخی موارد ممکن است داروها برای کنترل قسمت های ضربان قلب غیرطبیعی کافی نباشد و افراد ممکن است قادر به تحمل دارو باشند. در چنین مواردی ممکن است یک روش جراحی تحت عنوان تخلیه کاتتر شناخته شود. این روش همچنین می تواند در افرادی که در معرض خطر ابتلا به سرکوب قلبی و مرگ ناگهانی از جمله برخی از افراد بدون علامت هستند استفاده شود.
در طی یک تخلیه کاتتر، یک لوله نازک کوچک (کاتتر) به قلب وارد می شود و به مسیر غیر طبیعی هدایت می شود که در آن انرژی الکتریکی با فرکانس بالا برای تخریب (تخلیه) بافت ساختاری مسیر غیرطبیعی استفاده می شود. این فرم درمان دارای میزان موفقیت بسیار بالایی است و ممکن است در بسیاری از افراد نیاز به دارو داشته باشد.
در گذشته عمل جراحی قلب باز برای درمان افراد مبتلا به سندرم WPW مورد استفاده قرار گرفت. با توجه به موفقیت روش کمتر تهاجمی، کاتتر (رادیوفرکوسی) تخلیه، جراحی قلب باز به ندرت برای افراد مبتلا به سندرم WPW انجام می شود.