پیشینه
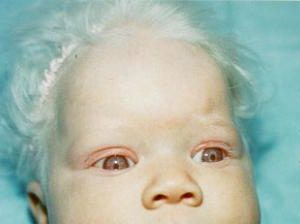
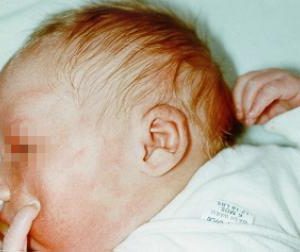
طبقه بندی بیماریهای کمبود مادرزادی رنگ دانهای (hypopigmentary) که ناشی از نقص در تولید رنگ دانه (ملانین) به علت اختلال در سلولهای تولید کننده رنگدانه (ملانوسیتها) در پوست، چشم و یا گوش است، شامل موارد زیر میباشد: آلبینیسم چشمی-صورتی (oculocutaneous) نوع 1-7؛ آلبینیسم چشمی؛ سندرم چدیاک-هیگاشی (Chediak-Higashi) که در تصویر زیر مشاهده میشود؛ سندرم هرمانسکی-پودلاک (Hermansky-Pudlak)؛ و سندرم گریسلی (Griscelli).
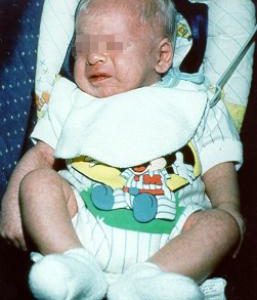
سندرم چدیاک-هیگاشی و سندرم هرمانسی-پودلاک همچنین با نقایصی غیر از مشکلات رنگ دانهای از جمله اختلال در لکوسیت، پلاکت، پنوموسیت و اختلال در سلولهای رتیکولار شناخته میشوند. همچنین سندرم گریسلی نیز میتواند با نقص ایمنی و اختلال عصبی همراه باشد.
پاتوفیزیولوژی
مشخصه این بیماریها کمبود کامل یا جزئی در رنگدانه پوست و مو میباشد. جهش در ژنهایی که تنظیم کننده روند چند مرحلهای سنتز ملانین، توزیع رنگدانه توسط ملانوسیت و/یا تولید ملانوزومها هستند، از دلایل اصلی بروز این بیماریها میباشند. محصولات پروتئین/ژن (و ژن مربوطه) در هر نوع آلبینیسم چشمی-پوستی تحت تاثیر قرار میگیرند در ادامه گفته شده است:
- آلبینیسم چشمی-پوستی نوع 1: آنزیم تیروزیناز [11q14-21]
- آلبینیسم چشمی-پوستی نوع 2: P پروتئین [15q11-13]
- آلبینیسم چشمی-پوستی نوع 3: آنزیم پروتئین 1 مرتبط با تیروزیناز (TRYP1) [9p23]
- آلبینیسم چشمی-پوستی نوع 4- نوعی پروتئین حامل مرتبط با غشا (MATP/SLC24A2) [5p13.3]
- آلبینیسم چشمی-پوستی نوع 5: پروتئین ناشناخته [4q24]
- آلبینیسم چشمی-پوستی نوع 6- نوعی پروتئین حامل مرتبط با غشا (SLC24A5) [15q21.1]
- آلبینیسم چشمی-پوستی نوع 7: پروتئین ناشناخته [10q22.2-3]
علل آلبینیسم
علل این بیماریها جهش در ژنهای به خصوصی میباشد.
- آلبینیسم چشمی-پوستی نوع 1 به علت جهش در ژن تیروزیناز، که در جایگاه 11q14-3 وجود دارد، به صورت یک صفت اتوزومی مغلوب به ارث میرسد. ژن تیروزیناز آنزیمی را کد میکند که سنتز ملانین را با استفاده از سوبسترای تیروزین آغاز میکند. به طور ویژه، تیروزیناز تیروزین را به دی هیدروکسی فنیل آلانین (DOPA) هیدروکسیله کرده و متعاقب آن DOPA را به DOPA-اکسیداز دهیدروکسیله مینماید. بیش از 70 جهش در آنزیم تیروزیناز شناسایی شده است که منجر به اختلال عملکرد یا سنتز کم این آنزیم میشوند. اکثر بیماران مبتلا به آلبینیسم چشمی-پوستی نوع 1 نسبت به جهش ژن تیروزیناز هتروزیگوت هستند.
- آلبینیسم چشمی-پوستی نوع 2 به علت جهش در Pgen، که در جایگاه 15q12 قرار دارد، به صورت یک صفت اتوزوم مغلوب به ارث میرسد. ژن P یک پروتئین 110 کیلو دالتونی کد میکند که؛ 12 جایگاه خلال غشایی محدود شده بر روی غشای گرانولهای حاوی ملانین (یعنی ملانوزوم) دارد. عملکرد پروتئین P در سنتز ملانین هنوز مشخص نشده است.
- آلبینیسم چشمی-پوستی نوع 3 به علت جهش در ژن پروتئین 1 مرتبط با تیروزیناز (Tyrp1)، که به جایگاه 9p23 متعلق است، به صورت یک صفت اتوزوم مغلوب به ارث میرسد. ژن Tyrp1 پروتئینی را کد میکند که به نظر میرسد در بدن موش فعالیت دی هیدروکسیاندول کربوکسیلیک اسید (DHICA) اکسیدازی دارد. DHICA اکسیداز یک مرحله کاتالیستی پایینتر از تیروزیناز در بیوسنتز ملانین از تیروزین است. در انسان عملکرد Tyrp1 طی فرآیند ساخت ملانین ممکن است به صورت (1) یک حامل یونی، (2) یک پروتئین چپرون (chaperone)، و/یا (3) تثبیت کننده کمپلکس ملانوزوم باشد.
- آلبینیسم چشمی-پوستی نوع 4 به علت جهش در ژن SLC45A2، که قبلا ژن پروتئین حامل مرتبط به غشاء (MATP) نامیده میشد و به جایگاه 5p13.3 متعلق است، به صورت یک صفت اتوزوم مغلوب به ارث میرسد. ژن SLC45A2 کد کننده یک پروتئین 58 کیلودالتونی با 12 قسمت پیش بینی شده خلال غشایی است. در حال حاضر عملکرد MATP در فرآیند ساخت ملانین ناشناخته است.
- آلبینیسم چشمی-پوستی نوع 5 به علت جهش در یک ژن ناشناخته که به جایگاه 4q24 متعلق است ایجاد شده و به صورت یک صفت اتوزومی مغلوب به ارث میرسد. خوده پروتئین و عملکرد آن نامعلوم است.
- آلبینیسم چشمی-پوستی نوع 6 به علت جهش در ژن SLC24A5، که به جایگاه 15q21.1 متعلق است، به صورت یک صفت اتوزوم مغلوب به ارث میرسد. ژن SLC45A5 یک پروتئین حامل مرتبط با غشای نامشخص را رمزگذاری میکند و عملکرد آن ناشناخته است.
- آلبینیسم چشمی-پوستی نوع 7 به علت جهش در یک ژن ناشناخته که به جایگاه 10q22.2-3 متعلق است، به صورت یک صفت اتوزومی مغلوب به ارث میرسد. پروتئین به طور موقت به عنوان C10orf11 نام گذاری شده و عملکرد آن نامعلوم است.
- آلبینیسم چشمی به علت جهش در ژنی در کروموزوم X که به جایگاه Xp22.3-22.2 مرتبط است ایجاد میشود و به صورت یک صفت وابسته به X مغلوب به ارث میرسد. عملکرد محصول ژن آلبینیسم چشمی شناخته نشده است.
- سندرم چدیاک-هیگاشی به علت جهش در ژن LYST، که به جایگاه 1q42-43 متعلق است، به صورت یک صفت اتوزوم مغلوب به ارث میرسد. ژن LYST یک پروتئین بزرگ 429 کیلو دالتونی را کد گذاری میکند که ظاهرا عملکرد آن در انتقال ماده از دستگاه گلژی به جایگاههای هدف در سلولهای آسیب دیده است. در نتیجه سنتز ملانوزومها توسط ملانوسیت، گرانولهای دلتا توسط پلاکت و لیزوزومها توسط برخی از لکوسیتها (یعنی نوتروفیلها و لنفوسیتهای کشنده طبیعی) کاهش مییابد.
- سندرم هرمانسکی-پودلاک به صورت یک صفت اتوزومی مغلوب به ارث میرسد و ناهمگونی لوکوس در این بیماری دیده میشود. شکل ابتدایی سندرم هرمانسکی-پودلاک شناسایی، به نام سندرم هرمانسکی-پودلاک نوع 1 نام گذاری شده است و عوارض بیماری به علت ژنی که در جایگاه 10q23.1-23.3 قرار دارد، حاصل میشود. تا به امروز، 8 گونه ژنتیکی متمایز از سندرم هرمانسکی-پودلاک در جمعیت انسانی شناسایی شده است. بیشتر محصولات ژن سندرم هرمانسکی-پودلاک برای تشکیل چندین کمپلکس که انتقال مولکولها از شبکه گلژی به اندامهای هدف را تسهیل میکند، ترکیب میشوند.
- سندرم گریسلی به صورت یک صفت اتوزوم مغلوب به ارث میرسد. دو فرم ژنتیکی اولیه شناخته شده است. یکی در نتیجه جهش در ژن RAB27A واقع در جایگاه 15q21 که پروتئین Rab27a متصل به GTP را کد میکند. دیگری در نتیجه جهش در ژن MYO5A واقع در جایگاه 15q21 که پروتئین میوزین حرکتی غیرمعمول myosin5a را رمز گذاری میکند. هر دو لوکوس ژنی از یکدیگر متفاوت هستند. در ملانوسیت، این 2 محصولات ژنی همراه با یک پروتئین ارتباط دهنده سوم (یعنی ملانوفیلین)، ترکیب پیچیدهای را ایجاد میکنند که انتقال ملانوزومها را در امتداد میکروتوبولها در دندریتهای ملانوسیت و احاطه آنها به وسیله رشتههای اکتین در نواحی دندریتیکی را تسهیل میکند.
اپیدمیولوژی آلبینیسم
شیوع
بروز تقریبی این بیماریها به صورت زیر است:
- آلبینیسم چشمی-پوستی نوع 1: یک مورد در هر 40000 نفر
- آلبینیسم چشمی-پوستی نوع 2: یک مورد در هر 360000 نفر، به جز در آفریقاییها و آمریکاییهای آفریقایی تبار، که در این جمعیت میزان بروز یک مورد در هر 10000 نفر است.
- آلبینیسم چشمی-پوستی نوع 3: نامعلوم
- آلبینیسم چشمی-پوستی نوع 4: یک مورد در هر 100000 نفر، به جز در ژاپن، که 24٪ از افراد با آلبینیسم چشمی-پوستی به این فرم مبتلا هستند.
- آلبینیسم چشمی-پوستی نوع 5: نامعلوم (تنها در یک خانواده گزارش شده است)
- آلبینیسم چشمی-پوستی نوع 6: نامعلوم (تنها در 2 نفر گزارش شده است)
- آلبینیسم چشمی-پوستی نوع 7: نامعلوم (تنها در چند نفر گزارش شده است)
- آلبینیسم چشمی: یک مورد در هر 50000 نفر
- سندرم چدیاک-هیگاشی: بسیار نادر
- سندرم هرمانسکی-پوداک: نادر است، به جز در پورتوریکو، که در این منطقه فراوانی بیماری 1 در هر 1800 نفر است.
- سندرم گریسلی: بسیار نادر
نژاد
به نظر میرسد همه نژادها به طور یکسان تحت تاثیر جهشهای مرتبط با بیماری میباشند. با این حال، آلبینیسم چشمی-پوستی نوع 2 در میان آفریقاییها و آمریکاییهای آفریقایی تبار (1 مورد در هر 10000 نفر) بیشتر از سفید پوستان (1 مورد در هر 36000 نفر) گزارش شده است.
جنسیت
احتمال بروز این بیماری در مردان و زنان برابر است.
سن
همه این بیماریها در نوزادان دیده میشوند. سندرم چدیاک-هیگاشی یک فاز شتابنده دارد که سالها تا دههها بعد از تولد بروز مییابد.
پیشآگهی آلبینیسم
آلبینیسم چشمی-پوستی نوع 1، 2، 3، 4 و آلبینیسم چشمی با مرگ و میر و یا بیماری به جز حساسیت پوستی به تابش و اشعه خورشید و نقصهای بینایی مرتبط با آن که در ادامه ذکر شده، مرتبط نیستند.
کودکان مبتلا به سندرم چدیاک-هیگاشی علائم کبودی آسان، خونریزی مخاطی، خون ریزی از بینی و پتشی، عفونتهای مکرری که اصولا سیستم تنفسی را درگیر میکنند، و نوتروپنی را تجربه میکنند. تقریبا 85٪ از افراد مبتلا به سندرم چدیاک-هیگاشی با ورود به یک مرحله شتابنده، با علائمی از جمله تب، کم خونی، نوتروپنی و گاهی اوقات ترومبوسیتوپنی، بزرگی طحال و کبد، لنفادنوپاتی، و زردی درگیر میشوند. مشکلات عصبی در سندرم چدیاک-هیگاشی متغیر بوده و نوروپاتی محیطی و اعصاب جمجمهای، نوروپاتی اتونومیک، ضعف و نارسایی حسی، از دست رفتن رفلکسهای عمیق تاندون، مشکل گسترده در طرز راه رفتن (gait)، تشنج، و کاهش سرعت هدایت عصب حرکتی را شامل میشود. مرگ معمولا در دهه اول پس از شروع عفونت، خونریزی یا بروز فاز تسریعی رخ میدهد.
افراد مبتلا به سندرم هرمانسکی-پودلاک به دلیل کمبود ذخیره پلاکت مستعد خونریزی هستند. همچنین این افراد به یک بیماری ذخیرهای سروئید مبتلا میشوند که در آن مواد سروئید-لیپوفوشین در سیستمهای مختلف بدن تجمع یافته و منجر به فیبروز ریوی، کولیت گرانولوماتوز، التهاب لثه (gingivitis)، نارسایی کلیه و كاردیومیوپاتی میشود. فیبروز ریوی معمولا در دهه چهارم یا پنجم زندگی کشنده است. نه شکل ژنتیکی مختلفی از سندرم هرمانسکی-پودلاک وجود دارد.
اکثر افراد مبتلا به سندرم گریسلی به عفونتهای مزمن مبتلا میشوند که ناشی از نقص شدید ایمنی است و در طول دهه اول زندگی میتواند کشنده باشد.
وراثت
بسته به علت ژنتیکی این بیماری، انواع مختلف آلبینیسم الگوهای وراثتی مختلفی میتوانند داشته باشند. آلبینیسم چشمی-پوستی (OCA) چشمها، مو و پوست را درگیر میکند. آلبینیسم چشمی (OA)، که کمتر رایج است، عمدتا چشمها را درگیر میکند، در حالی که ممکن است پوست و مو این افراد مشابه یا کمی روشنتر از سایر اعضای خانواده باشد. جهشها در چندین ژن مختلف، بر روی کروموزومهای متفاوت، میتواند انواع مختلف آلبینیسم را ایجاد کند.
OCA به صورت صفت اتوزومی مغلوب به ارث میرسد. به این معنی که در هر فرد وجود دو جهش برای بروز OCA ضروری است. این افراد معمولا دو کپی از هر کروموزوم گفته شده و ژنهای روی آنها را دارند- یکی از پدر و دیگری از مادر به ارث رسیده است. هیچ کدام از این نسخههای ژنی در افراد مبتلا به آلبینیسم عملکرد صحیح ندارند. هر یک از پدر و مادر سالم فرد مبتلا به بیماری اتوزومی مغلوب، یک نسخه عملکردی سالم از ژن مسبب بیماری و یک نسخه غیرعملکردی دارند. در واقع آنها به عنوان فرد حامل شناخته میشوند و معمولا نشانهها یا علائم بیماری را نشان نمیدهند. هر دو والد باید یک نسخه معیوب از ژن OCA داشته باشند تا یک کودک مبتلا به آلبینیسم متولد شود. هنگامی که هر دو فرد برای یک صفت اتوزوم مغلوب حامل باشند، در هر حاملگی 25٪ (1 در 4) خطر بیمار بودن کودک، 50٪ (1 در 2) خطر حامل بودن کودک مانند هر یک از والدین بدون بروز علائم بیماری، و یک احتمال 25 درصدی وجود دارد که کودک نه بیمار باشد و نه حامل.
آلبینیسم چشمی نوع 1 با الگوی وابسته به X به ارث میرسد. بیماری در صورتی وابسته به X تلقی میشود که ژن جهش یافته مسبب بیماری بر روی کروموزوم X، یکی از دو کروموزوم جنسی، واقع شود. در مردان (که فقط یک کروموزوم X و یک کروموزومY دارند) وجود یک کپی تغییر یافته از ژن عامل بیماری در هر سلول برای بروز ویژگیهای مشخص آلبینیسم چشمی کافی است، زیرا مردان کروموزوم X دیگر با ژنهای عملکردی ندارند. از آن جا که زنان دو نسخه از کروموزوم X دارند، زنانی که در هر سلول تنها یک کپی از ژن جهش یافته را دارا باشند، معمولا کاهش بینایی و یا سایر اختلالات بینایی قابل ملاحظهای تجربه نمیکنند. تنها ممکن است تغییرات خفیفی در رنگدانه شبکیهای داشته باشند که این تغییرات در طول معاینات چشم فرد قابل شناسایی هستند.
محققان همچنین چندین ژن دیگر را شناسایی کردهاند که جهشهای ایجاد شده در آنها میتواند باعث ایجاد آلبینیسم همراه با سایر ویژگیها شود. یک گروه از این تغییرات شامل حداقل نه ژن (بر روی کروموزومهای مختلف) است که منجر به سندرم هرمانسکی-پوداک (HPS) میشود. علاوه بر آلبینیسم، HPS با اختلالات خونریزی و کبودی همراه است. بعضی از اشکال نیز با بیماری ریوی و رودهای مرتبط هستند. مانند OCA ،HPS نیز به صورت اتوزومی مغلوب به ارث میرسد.
درمان
هیچ درمانی برای هيپوپيگمانتاسيون پوست، مو يا چشم در دسترس نيست. برای جلوگیری از آسیبهای ناشی از اشعه ماوراء بنفش به پوست استفاده از کرمهای ضد آفتاب وسیع الطیف (broad-spectrum) و پوشیدن لباس توصیه میشود. اختلالات بینایی را میتوان با استفاده از لنزهای اصلاح شده بهبود داد.
بیشتر برای سندرم چدیاک-هیگاشی و سندرم گریسل درمان علامتی را به کار میبریم. برای درمان عفونتها آنتیبیوتیکهای مناسب باید تجویز شوند. پیوند مغز استخوان میتواند به ترتیب نقایص هماتولوژیکی و ایمونولوژیکی را در افراد مبتلا به سندرم چدیاک-هیگاشی و سندرم گریسلی اصلاح و بهبود بخشد.
هیچ روش درمانی برای اختلالات غيرپيگمانتاسيون سندرم هرمانسکی-پودلاك موثر نيست. اگر فرد مستعد خونریزی شدید باشد، انتقال پلاکت و خون ممكن است مفید واقع شود. اگر کولیت گرانولوماتوزی یا فیبروز ریوی تشدید شود، ممکن است تجویز استروئید با دوز بالا برای فرد در نظر گرفته شود.