اختلال استخوان زایی یا استئوژنز ایمپرفکتا (Osteogenesis imperfecta) نوعی اختلال استخوانی است که در اثر جهش در ژنهای COL1A1 و COL1A2 بروز پیدا کرده و با شکنندگی افزایش یافتۀ استخوان همراه است. پروتئین حاصل از بیان این دو ژن، پروکلاژن نوع یک نام دارد. چهار نوع متفاوت از اختلال استخوان زایی در سال ۱۹۷۹ توسط سیلنس (Sillence) تعریف شد، که امروزه همگی در قالب “معیارهای سیلنس” مورد پذیرش قرار گرفتهاند. در ویرایش مجدد سال ۲۰۱۰ “بیماریشناسی و طبقهبندی اختلالات ژنتیکی دستگاه اسکلتی” نیز طبقهبندی مشابهی از انواع اختلال استخوان زایی ارائه شده است. تشخیص نوع اختلال اغلب دشوار بوده و تا حد زیادی به تخصص بالینی پزشک بستگی دارد. شدت بیماری از انواع خفیف تا موارد بسیار شدید در دوران پیرازایشی، متغیر است. ژنهای جدیدی شناسایی شدهاند که جهش در آنها نیز به رشد استخوانهای ضعیف و به نسبت شکننده میانجامد. چنین اختلالاتی در اکثر مواقع غیرقابل تمییز بوده، و از سوی جامعه پزشکی به عنوان زیردستههای استئوژنز ایمپرفکتا شناخته میشوند. نمونههایی از یافتههای متداول در رادیولوژی اختلال استخوان زایی در تصاویر زیر آورده شده است.
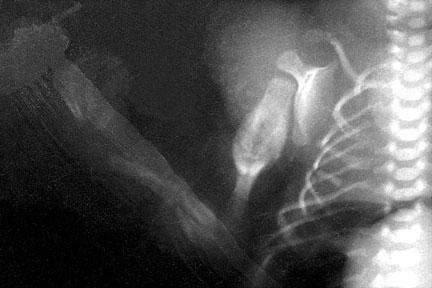
شکستگیهای حاد در استخوانهای رادیوس و اولنا قابل مشاهده است. چندین مورد شکستگی در دندهها دیده میشود. پینههای واقع در محل شکستگیهای قدیمی استخوان بازو نیز مشاهده میگردد.
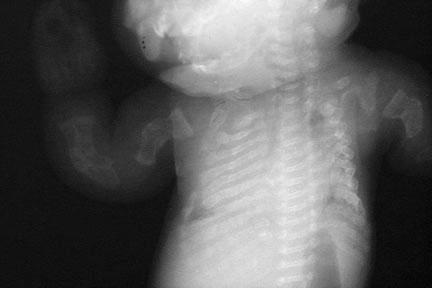
خوردهشدگی دندهها. چندین نمونه شکستگی در اندام فوقانی نیز مشاهده میگردد.
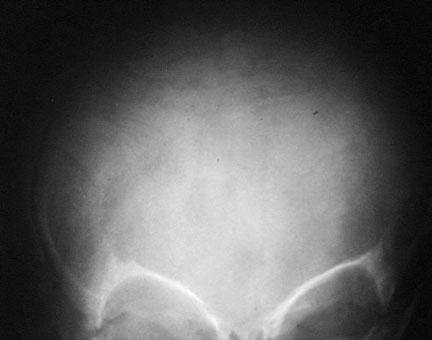
استخوانهای بین درزی در جمجمه.
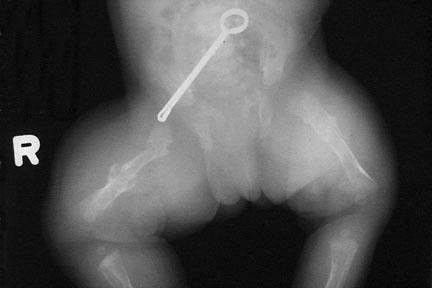
شکستگی دو طرفه استخوان ران.
پاتوفیزیولوژی
پرکاربردترین طبقهبندی از انواع اختلال استخوان زایی که در سال ۱۹۷۹ توسط سیلنس و همکارانش ارائه گردید، انواع جدیدتر این اختلال را شامل نمیشود؛ که به لطف پیشرفتهای صورت گرفته در حوزۀ تشخیص مولکولی، بعدها توسط دانشمندان شناسایی شدند. انواع کمتر شناخته شده از استئوژنز ایمپرفکتا، اغلب در نتیجۀ بروز جهش در ژنهای مرتبط با ساخته شدن کلاژن نوع یک، و یا ژنهای دخیل در استخوانسازی، ناشی میگردد. این زیردستههای تازه کشف شده، از نظر خاستگاه مولکولی با انواعی از اختلال استخوان زایی که با ژنهای معیوب COL1A1 و COL1A2 در ارتباطاند، تفاوت داشته، و نمونهای از ناهمگونی در جایگاه ژنی (locus heterogeneity) به شمار میآیند. در سال ۲۰۱۵، فورلینو و مارینی، طبقهبندی جدیدی را به منظور درک بهتر اساس ژنتیکی اختلال استخوان زایی ارائه دادند، که از پنج دستۀ عملکردی جداگانه تشکیل شده است:
- دستۀ A: نواقص اولیه در ساختار یا عملکرد کلاژن (COL1A1، COL1A2 و BMP1)
- دستۀ B: نقص در پردازش پساترجمهای کلاژن (CRTAP، LEPRE1، PPIB، TMEM38B)
- دستۀ C: نقص در تاخوردگی و اتصالات متقاطع بین رشتههای کلاژن (SERPINH1، FKBP10، PLOD2)
- دستۀ D: نقص در معدنیسازی و استخوانی شدن (IFITM5 و SERPINHF1)
- دستۀ E: نقص در نمو استئوبلاست به همراه نارسایی کلاژن (WNT1، CREB3L1، SP7)
این دستگاه طبقهبندی هنوز کاربرد چندانی ندارد، ولی انواع اختلال استخوان زایی را به شکلی بسیار ساده و قابل فهم از یکدیگر تفکیک کرده است.
COL1A1/COL1A2 (انواع I تا IV)
الیاف کلاژن نوع یک در ساختمان استخوان، کپسولهای پوشانندۀ اندامهای داخلی، فاسیا، قرنیه، صلبیه، تاندون، مننژ و درم، یافت میشود. کلاژن نوع یک، که تقریباً ۳۰ درصد از وزن بدن را تشکیل میدهد، در اختلال استخوان زایی دچار نقص میگردد. ۹۰ درصد از پروتئین موجود در ساختمان استخوان به کلاژن نوع یک تعلق دارد. از نظر ساختاری، الیاف کلاژن نوع یک از یک مارپیچ چپگرد ساخته شده است. این مارپیچ از زنجیرههای در هم تنیدۀ پرو-آلفا ۱ و پرو-آلفا ۲ تشکیل میشود. بروز جهش در ژنهای بیانکنندۀ این دو زنجیره میتواند به ظهور اختلال استخوان زایی بینجامد. چنین جهشهایی اغلب دو ژن COL1A1 و COL1A2 را در جایگاههای 17q21 و 7q22.1 متأثر میسازد.
نواقص کیفی (نظیر ناهنجاری در مولکولهای سازندۀ کلاژن I) و نواقص کمی (مانند تولید کاهش یافتۀ مولکولهای کلاژن I طبیعی) نیز در پاتوفیزیولوژی این اختلال توصیف شدهاند. نواقص کیفی در اکثر مواقع از جهشهای بدمعنی در کدونهای رمزگردان گلیسین ناشی گردیده، و به دلیل اثر منفی غالبی که چنین جهشهایی اعمال میکنند، منجر به بروز بیماری میشوند. همبستگی واضحی بین ژنوتیپ و فنوتیپ دیده نمیشود، اما مشاهداتی در این باره صورت گرفته است.
نخست، بروز جهش در ژن COL1A1 با پیامدهای شدیدتری نسبت به ژن COL1A2 همراه است. این میتواند به این دلیل باشد که برای ساخته شدن هتروتریمر پروکلاژن نوع یک، دو زنجیره آلفا ۱ و تنها یک زنجیرۀ آلفا ۲ مورد نیاز است. دوم، جهشهای واقع در انتهای کربوکسیلی هر یک از ژنها، نسبت به بقیۀ دومِینها، با پیامدهای بارزتری همراهاند. به ویژه، جهشهای بدمعنی در دومین رمزگردان انتهای کربوکسیلی COL1A1، اغلب (ولی نه همیشه) کشندهاند. سوم، تغییرات صورت گرفته در اندازه و قطبیت مولکول حاصل از بیان ژن، در شدت عوارض ناشی از جهش، تأثیرگذار است. برای مثال، جایگزین شدن گلیسین با آلانین، که تنها اندکی بزرگتر است و خصوصیات شیمیایی یکسانی دارد، با پیامد خفیفی همراه بوده و چندان در فنوتیپ ظاهر نمیشود. در مقابل، جایگزین شدن گلیسین با گلوتامیک اسید در همان جایگاه قبلی، به دلیل بزرگی و بار منفی این آمینو اسید و خصوصیات شیمیایی متمایز آن، نمود بیشتری در فنوتیپ داشته و با علائم شدیدتری همراه است، به طوری که حتا ممکن است کشنده باشد. نواقص کمی اغلب از جهشهای بیمعنی ناشی میگردند، ولی گاهی ممکن است چنین نواقصی در اثر حذفشدگیهای بزرگتر در یکی از ژنهای رمزگردان پروکلاژن، در نتیجۀ نارسایی هاپلوتیپ به بروز بیماری بینجامد. mRNA حاصل از این ژن به طور غیرعادی کوتاهتر خواهد بود، که خود باعث واپاشی ناشی از جهش بیمعنی (NMD) شده، و در نهایت بیمار تنها خواهد توانست نیمی از کلاژن I مورد نیاز خود را بسازد؛ ولو این که از نظر جسمانی عادی به نظر برسد. چنین بیمارانی به اختلال استخوان سازی خفیف نوع ۱ مبتلا میشوند، که معمولاً باعث بدشکلی نمیگردد.
مطالعهای که توسط بالاسوبرامانیان و همکارانش انجام گردید، نشان داد که جهشهای COL1A1/COL1A2 به طور یکنواختی به تغییر در قطر الیاف کلاژن میانجامد.
اختلال استخوان زایی با کلسیفیکاسیون غشاهای بین استخوانی (نوع V)
بیماران مبتلا به این نوع از استئوژنز ایمپرفکتا به طور معمول شدت متوسطی از بیماری را تجربه میکنند، ولی به طور مکرر، در پی بروز شکستگی در تنۀ استخوانهای دراز یا انجام جراحیهایی که نیاز به استئوتومی دارند، پینههای هیپرپلاستیک در استخوانهایشان شکل میگیرد. این پینهها ممکن است تا سالها شکل و اندازۀ ثابتی داشته باشند، و یا این که دورههایی از رشد سریعتر را تجربه کنند. با این حال در موارد اندک، پیچخوردگی پینهها را میتوان انتظار داشت. بیماران همچنان به طور مکرر دچار کلسیفیکاسیون در غشای بین استخوانی ساعد و دررفتگی سر استخوان رادیوس میشوند؛ که با دشواری در چرخانیدن ساعد به داخل و خارج همراه است. همچنین احتمال دارد رادیودانسیته متافیزیال و سابفیزیال در رادیوگرافی این بیماران مشاهده شود. این وضعیت در نتیجۀ بروز جهشهای هتروزیگوت در ژن IFITM5 ناشی میشود. توارث آن از نوع اتوزوم غالب است.
بررسی مقاطع بافتی از استخوان در بیماران مبتلا به نوع V، حاکی از این بود که تیغههای استخوانی بدون نظم طبیعی در کنار یکدیگر قرار گرفته، و در بعضی از موارد، شبیه به یک تورینه به نظر میرسند؛ که در تضاد با چیدمان موازی طبیعی است که در مبتلایان به اختلال استخوان زایی دیده میشود.
شکلهای دیگر از اختلال استخوان زایی
SERPINFI (نوع VI)
بیماران مبتلا به این نوع از اختلال استخوان زایی اغلب شدت متوسطی از بیماری را تجربه میکنند. مشاهده شده است که این بیماران در هنگام تولد هیچگونه شکستگی در بدن خود ندارند، و بعدها در دوران شیرخواری یا پس از آن دچار شکستگی میشوند. رنگ صلبیه در این گروه از بیماران تا حدودی آبی و یا سفید بوده و دندانهای طبیعی دارند. این وضعیت در اثر بروز جهش هوموزیگوت در ژن SERPINF1 عارض میگردد و توارث آن از نوع اتوزوم مغلوب است.
تیغههای استخوانی در این بیماران نمایی شبیه به فلس ماهی داشته، و حداقل یک پژوهشگر به وجود مقادیر زیادی از استئویید معدنی نشده در بافت استخوان اشاره کرده است.
CRTAP/LEPRE1/PPIB (انواع VII تا IX)
بیماری ناشی از جهش در این سه ژن، اغلب با شدت زیادی همراه بوده و گاه کشنده است. پروتئین مرتبط با غضروف (CRTAP)، نوعی پروتئین میباشد که برای ۳ – هیدروکسیلاسیون پرولیل ضروری است. CRTAP در کنار محصول دو ژن LEPRE1 و PPIB، نوعی پروتئین هتروتریمریک (سه زیرواحد ناهمسان) میسازد که برای پیرایش پساترجمهای طبیعی کلاژن I ضروری است. اختلال استخوان زایی ناشی از جهش در CRTAP، نوع VII به حساب میآید. جهش در ژنهای LEPRE1 و PPIB نیز به ترتیب موجب ظهور استئوژنز ایمپرفکتای نوع VIII و IX میشود. این وضعیتها در نتیجۀ بروز جهشهای هوموزیگوس یا هتروزیگوس ترکیبی ظاهر شده، و نوع توارث آن نیز اتوزوم مغلوب است.
SERPINH1 (نوع X)
در متون تنها به یک مورد مبتلا به این نوع از اختلال استخوان زایی اشاره شده است. این بیمار مرد، حاصل ازدواج فامیلی مرد و زنی از عربستان سعودی بوده است. بنا بر گزارشها، والدین هر دو از نظر بالینی، طبیعی بودند. گزارش شده است که وی از نوع شدیدی از استئوژنز ایمپرفکتا رنج میبرده که باعث بدشکلی نیز میشده است. علائم همراهی نظیر اختلال در دندان زایی، رسوب کلسیم در کلیه (نفروکلسینوز) و بیماری مزمن ریوی نیز در این بیمار گزارش شده است. او در ۳ سالگی به دلیل دشواری در تنفس درگذشت. ارزیابی ژنتیک این کودک حاکی از وجود نوعی جهش هوموزیگوس در ژن SERPINH1 بود. پروتئین حاصل از بیان این ژن، یک چاپرون است که در شبکه اندوپلاسمیک مستقر شده و نقل و انتقال کلاژن را مدیریت میکند.
FKBP10 (نوع XI)
شمار اندکی از بیماران مبتلا به اختلال استخوان زایی علائم مربوط به این دسته را نشان میدهند. شدت این نوع از بیماری متوسط تا شدید ارزیابی شده است. استئوژنز ایمپرفکتای نوع XI در اثر بروز جهش هوموزیگوس در ژن FKBP10 ایجاد شده، و توارث آن نیز از نوع اتوزوم مغلوب است. گزارش شده است که تعدادی از بیماران مبتلا به نوع II سندرم براک (Bruck syndrome)، دارای جهش در این ژن هستند. با این حال، این طیف از یافتههای بالینی هنوز به خوبی درک نشده است. پیشنهاد شده که اختلالات مربوط به FKBP10 جزو انواع مغلوب و پیشرونده از اختلال استخوان زایی طبقهبندی شود، که ممکن است با شکستگی همراه باشند و یا نباشند. پروتئین حاصل از بیان FKBP10، یک چاپرون است که در فرآیند تاخورده شدن کلاژن شرکت میکند.
بافتشناسی استخوان نشان دهنده اعوجاج در چیدمان تیغههای استخوانی و الگوی فلس ماهی است.
SP7 (نوع XII)
فقط یک بیمار مبتلا به این وضعیت گزارش شده، و بیماری وی شدت متوسطی داشته است. این بیمار مذکر حاصل ازدواج بین دو عموزاده درجه دو بود، که در کنار شکستگی، دچار تأخیر در جوانهزنی دندان نیز بوده است. این بیمار شنوایی طبیعی و صلبیۀ سفید داشته است. یکی دیگر از فرزندان این خانواده نیز با همین اختلال به دنیا آمده بود، و نهایتاً در اثر نقص مادرزادی قلب – که ظاهراً ارتباطی به این اختلال نداشت – درگذشت. ارزیابی ژنتیک این بیمار حاکی از حذفشدگی هوموزیگوس در ژن SP7 بود. این نوع از استئوژنز ایمپرفکتا توارث اتوزوم مغلوب دارد. محصول ژن SP7 برای تمایز استئوبلاستها و ساخته شدن استخوان ضروری است. در مدل حیوانی فاقد این ژن، هیچگونه استخوان قشری یا تیغۀ استخوانی از طریق استخوانی شدن داخل غشایی تشکیل نشد.
BMP1 (نوع XIII)
تعداد کمی از بیماران مبتلا به این نوع از استئوژنز ایمپرفکتا گزارش شدهاند. این بیماران علائم شدید به همراه شکستگیهی متعدد و اختلال رشد داشتند. با این حال، دو برادر مبتلا به این بیماری تنها از کمتراکمی مرزی استخوان رنج میبردند. بیماران این دسته دندانهای طبیعی و صلبیۀ آبیرنگ دارند. اختلال استخوان زایی نوع XIII در نتیجۀ بروز جهش هوموزیگوس در ژن BMP1 ظاهر شده، و نوع توارث آن نیز اتوزوم مغلوب است. پروتئین BMP1 نقش مهمی در استخوان زایی دارد. این پروتئین همچنین در حذف C – پروپپتیدها از انواع خاصی از پروکلاژنها، به خصوص پروکلاژن I، نقش ایفا میکند.
TMEM38B (نوع XIV)
این نوع از استئوژنز ایمپرفکتا فرزندان حاصل از سه مورد ازدواج فامیلی در دو کشور عربستان سعودی و اسرائیل گزارش شده است. بیماران شدت متغیری از بیماری را تجربه کرده، و صلبیۀ آبی، اختلال دندان سازی و ناشنوایی در آنها مشاهده نمیشود. این نوع از اختلال استخوان سازی ناشی از بروز جهت هوموزیگوس در ژن TMEM38B بوده، و به صورت اتوزوم مغلوب به ارث میرسد.
WNT1 (نوع XV)
این نوع تازه کشف شده از اختلال استخوان سازی در تعدادی از خانوادهها توصیف شده، و از شدت متوسط تا بالا برخوردار است. بیماران مبتلا به این وضعیت قامت کوتاه، صلبیۀ آبی یا سفیدرنگ و شنوایی طبیعی دارند. بدشکلیهای مغزی و تأخیر در نمو، در یک زیردسته از بیماران گزاراش شده است. جهش هوموزیگوس در ژن WNT1، که به صورت اتوزوم مغلوب به ارث میرسد، به ظهور این بیماری منجر میشود. مسیر سیگنالینگ WNT/beta-catenin اهمیت وافری در تمایز استئوبلاستها و تکامل استخوان دارد.
CREB3L1 (نوع XVI)
CREB3L1 پروتئین مبدل استرس OASIS را در دستگاه اندوپلاسمیک کد میکند. این پروتئین نیز در ادامه بیان پروکلاژن نوع یک را تنظیم میکند. جهش در هر دو آلل این ژن میتواند به نارسایی در تولید پروکلاژن ۱ بینجامد. گزارش شده است که دو نفر از فرزندان یک خانواده به این نوع از اختلال استخوان زایی مبتلا بودهاند. این دو بیمار هر دو جهش هوموزیگوس از نوع حذفشدگی در کروموزومشان داشتند. این حذف در جایگاهی به طول ۹۱ کیلوباز رخ میدهد. اولین بیمار مبتلا به نوع XVI که شناسایی گردید، مرد بود و از بیماری شدیدی نیز رنج میبرد. این بیمار جثۀ کوچکی در حین تولد داشت و متحمل چندین شکستگی در دورۀ جنینی شده بود. او پس از تولد دچار چندین شکستگی دیگر و خوردگی استخوانهای دنده شد. این بیمار به ذاتالریه نیز مبتلا بود و سرانجام در ۹ ماهگی، ظاهراً به دلیل دشواری در تنفس، جان خود را از دست داد. برادر وی نیز به همین شکل بیماری مبتلا بود، و در ۱۹ هفتگی سقط شد. والدین این دو نوزاد، و خواهر یکی از والدها، فاقد هرگونه شکستگی در استخوان بودند، اما رنگ صلبیۀ آنها آبی گزارش گردید. پدر دو بیمار پوست مخملی داشت و به ناشنوایی هدایتی مبتلا بود.
SPARC (نوع XVII)
SPARC یک گلیکوپروتئین اسیدی سرشار از سیستئین است که به چندین مورد از پروتئینهای مادۀ زمینهای، از جمله کلاژن نوع یک، اتصال مییابد. گزارش شده است که دو بیمار (غیرخویشاوند) واریانتهای غیرطبیعی از ژن SPARC را در ژنوم خود داشتند. هر دو بیمار از بیماری شدید رنج میبردند. بیماری این دو نفر در آن زمان، با توجه به فنوتیپ، اختلال استخوان زایی نوع IV تشخیص داده شده بود. بیمار اول اصالتاً اهل آفریقای شمالی بود. وی جدا از شکستگی استخوان، پس از تولد دچار خونریزی داخل بطنی شده بود. بیمار با آن که ۱۰ مورد شکستگی در استخوانهای دراز را از سر گذرانیده بود، ولی قامتش طبیعی به نظر میرسید. او دارای مفاصل بیش از حد انعطافپذیر و خمشدگی استخوان ران بود، و همچنین نسبت به سن خود، تراکم کمی از مواد معدنی در استخوانهایش داشت.
بیمار دوم حاصل ازدواج فامیلی والدین هندی بود. پزشکان در ابتدا دررفتگی مفصل لگن را در وی گزارش کرده، و سپس پی بردند که او دچار هیپوتونی و تأخیر در حرکت است. این بیمار قامت طبیعی، مفاصل انعطافپذیر و پوست نرمی داشت. تراکم مادۀ معدنی استخوان نسبت به سن بیمار کم ارزیابی شد. برای رسیدن به تشخیص درست، توالییابی کل اگزوم (WES) از هر دو بیمار به عمل آمد. بیمار اول برای واریانت غیرطبیعی c.497G>A هوموزیگوت بود. بیمار دوم نیز دو نسخه از واریانت غیرطبیعی c.787G>A را در ژنوم خود داشت. مشاهده گردید که واریانتهای بیماریزا، بخشهای به شدت محافظت شده از پروتئین را تحت تأثیر قرار میدادند. بررسیهای بیشتر نشان داد که این ناهنجاریها در بخش خارج سلولی اتصالیابنده به کلاژن پروتئین SPARC قرار گرفته بود.
اختلال استخوان زایی با همکشش مادرزادی مفاصل، نوع یک و دو (سندرم براک)
بیماران مبتلا به سندرم براک از همان ابتدای تولد استخوانهای ضعیفی دارند که مستعد شکستگی است. آنها علاوه بر این مشکل، دچار همکششی مفاصل و ناخنک چشم نیز هستند. کوتاهی قامت، بدشکلی شدید اندامها، وجود استخوانهای ورمیان یا بین درزی در جمجمه، و اسکولیوز پیشرونده از جمله سایر تظاهرات سندرم براک به شمار میروند. این بیماری شنوایی طبیعی و صلبیۀ سفیدرنگ داشته و اختلال دندان زایی ندارند. نتایج آزمایشها حاکی از وجود دو نوع متمایز از سندرم براک است؛ ولی این دو نوع در بالین قابل افتراق نیستند. سندرم براک ۱ در اثر بروز جهش هوموزیگوس در ژن FKBP10 ایجاد میشود، حال آن که نوع ۲ این سندرم از جهشهای هوموزیگوت در ژن PLOD2 ناشی میگردد. هر دو اختلال توارث اتوزوم مغلوب دارند. گفته میشود علت اصلی بروز این سندرم، وجود نقص در آنزیم مختص به استخوان تلوپپتید لیزیل هیدروکسیلاز است. آنزیم ناکارآمد باعث ایجاد اتصالات متقاطع غیرطبیعی بین الیاف کلاژن میشود.
جمعیتشناسی
اختلال استخوان زایی هر دو جنس را به یک اندازه گرفتار میکند. شمار دقیق بیماران مبتلا به این اختلال در ایالات متحده نامعلوم است. نوع I در یک مورد از هر ۳۰ هزار تولد زنده دیده میشود. طبق برآوردها، نوع II یک نفر از هر ۶۰ هزار تولد زنده را متأثر میسازد. شیوع کلی تمام انواع استئوژنز ایمپرفکتا در ۰.۵ مورد از هر ۱۰ هزار تولد زنده در ایالات متحده دیده میشود. حدوداً ۲۰ تا ۵۰ هزار بیمار مبتلا به اختلال استخوان زایی در ایالات متحده زندگی میکنند.
توارث
الگوی توارث بسیاری از موارد اختلال استخوان زایی از نوع اتوزوم غالب است. این یعنی داشتن تنها یک نسخه از ژن معیوب در سلول برای بروز علائم بیماری کافی است. بسیاری از بیماران مبتلا به انواع I یا IV اختلال استخوان زایی جهش بیماریزا را از یکی از والدین خود به ارث میبرند. اکثر نوزادانی که به انواع شدید از بیماری (نوع II و III) دچار هستند، هیچگونه سابقۀ قبلی از آن در خانوادۀ خود ندارند. علت بروز بیماری در این نوزادان، جهشهای تکگیر یا جدید در ژنهای COL1A1 و COL1A2 است.
با شیوع کمتر، اختلال استخوان زایی الگوی توارث اتوزوم مغلوب نیز دارد. در این نوع از توارث، برای این که علائم بیماری بروز یابد، نیاز به دو کپی از ژن معیوب در هر سلول است. والدین این بیماران ظاهراً سالماند، اما هر کدام حامل یک نسخه از ژن معیوب هستند. تعدادی از موارد استئوژنز ایمپرفکتای نوع III، چنین توارثی را نشان میدهند. چنین مواردی از بیماری در اکثر اوقات از جهشهایی غیر از دو ژن COL1A1/COL1A2 ناشی میگردد. بیماری ناشی از جهش در ژنهای CRTAP و P3H1 نیز توارث اتوزوم مغلوب دارد.
سببشناسی
استئوژنز ایمپرفکتا نوعی اختلال ارثی است. نوع توارث انواع I تا V بیماری از نوع اتوزوم غالب بوده، و بیشتر اوقات از جهشهای غالب جدید ناشی میگردد. این الگوی توارث مسئول بروز ۹۰ تا ۹۵ درصد از موارد بیماری است. ۵ تا ۱۰ درصد باقیمانده از بیماران به صورت اتوزوم مغلوب و از والدین حامل ولی به ظاهر سالم خود بیماری را دریافت کردهاند.
عدهای موزاییسم سلولهای زایا را به عنوان توجیهی برای تولد بیش از یک فرزند مبتلا به بیماری در خانوادههای با والدین سالم مطرح کردهاند. در پارهای از موارد، موازییسم سوماتیک در والدینی که چندین فرزند مبتلا به انواع غالب از اختلال استخوان زایی را به دنیا آوردهاند، گزارش شده است.
درمان
درمان اختلال استخوان زایی بر روی علائم خاصی متمرکز است که در هر بیمار به چشم میخورد. هدف از درمان، پیشگیری از بروز علائم، حفظ توانایی حرکت بیمار، و تقویت استخوانها و عضلات است.
ورزش و درمانهای فیزیکی نقش مؤثری در تقویت عضلات، افزایش ظرفیت تحمل وزن و کاهش آسیبپذیری نسبت به شکستگی داشته است. درمان فیزیکی در آب (آبدرمانی) نیز کمککننده است، چرا که حرکت در آب خطر بروز شکستگی را کاهش میدهد. بیماران مبتلا به اختلال استخوان زایی باید با پزشک و درمانگر خود مشورت کرده و برنامۀ بیخطری برای درمان پیریزی کنند.
در یک رویکرد درمانی، میلههای فلزی از طریق جراحی در تنۀ استخوانهای دراز کاشته میشوند تا خطر شکستگی کاهش بیاید. آتلهای پلاستیکی به عنوان ابزار محافظتی که امکان آزادی حرکت را هم برای بیمار فراهم میکنند، جایگزین کستهای پلاستیکی شدهاند. لباسهای بادکردنی نیز میتوانند از کودکان خردسال در برابر آسیبهای احتمالی محافظت نمایند.
ممکن است جراحی برای بیماران با بدشکلیهای شدید استخوانی در ستون مهرهها یا در رفتگی مهره اول گردنی، ضروری باشد. رویکردهای درمانی برای ترمیم ناهنجاریهای دندان نیز ممکن است به بیمار کمک کند. افراد مبتلا، به خصوص بزرگسالان، باید از نظر سلامت شنوایی مورد معاینه قرار بگیرند. مشاوره ژنتیک میتواند برای بیماران و خانوادههایشان سودمند باشد. سایر روشهای درمانی از نوع حمایتی بوده و از شدت علائم میکاهند.