تاریخچه
در سال 1942 کلاین فلتر، گزارشی را ارائه داد که در آن، 9 نفر مرد را با ویژگیهای خاصی توصیف کرده بود: دیسژنزی بیضهها، آلت تناسلی کوچک، اختگی، ژنیکوماستی (بزرگی پستان)، افزایش میزان هورمونهای جنسی و آزواسپرمی (نبود اسپرم در مایع منی). علت وجود این علائم، اختلال اندوکرینی تصور میشد تا این که در سال 1959 جیکوبز کشف کرد که سندرم کلاین فلتر یک اختلال کروموزومی است که در آن یک کروموزوم X اضافی در ژنوم فرد وجود دارد که منجر به ظهور کاریوتایپ 47,XXY میشود.
امروزه سندرم کلاین فلتر (KS) را به جمعی از اختلالات کروموزومی نسبت میدهند که در آن، کاریوتایپ طبیعی جنس مرد 46,XY دارای حداقل یک کروموزم X اضافی است. آناپلوئیدی XXY، شایعترین اختلال کرورموزم جنسی انسان است که میزان شیوع آن یک نفر به ازای پانصد مرد است. همچنین، این سندرم، شایعترین عامل هیپوگنادیسم و ناباروری مردان است.
سایر آناپلوئیدیهای کروموزوم جنسی جزو گروه اختلالات کروموزومی کلاین فلتر هستند. نوع نادر این اختلال به صورت 48,XXYY و 48,XXXY در یک از هفدههزار تا پنجاههزار تولد جنس مذکر اتفاق میافتد. در حالی که شیوع 49,XXXXY یک نفر به ازای هشتادوپنجهزار تا صدهزار تولد جنس مذکر است.
سندرم کلاین فلتر با هیپوگنادیسم (بیضههای کوچک، کمبود یا نبود اسپرم)، ژنیکوماستی در بلوغ دیررس (بزرگی پستان)، شفاف و فیبروزه شدن لولههای سمینی فروس، افزایش سطح هورمونهای جنسی و اختلالات رفتاری مشخص میشود. ویژگیهای فنوتایپی سندرم کلاین فلتر در تصاویر نشان داده شده است:
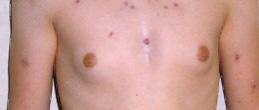
ژنیکوماستی در مرد بالغ مبتلا به سندرم کلاین فلتر
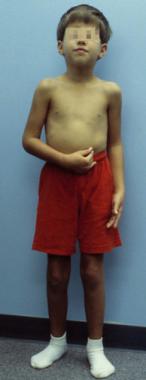
کودک مبتلا به سندرم کلاین فلتر. به جز ساختار استخوانی نازک و دستها و پاهای دراز و نامتناسب، فنوتیپ نرمال دارد.
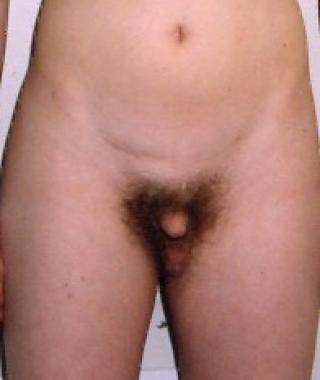
مرد بالغ مبتلا به سندرم کلاین فلتر که توزیع موی ناحیهی پوبیک مثل جنس مونث است و بیضهها رشد نکردهاند.
سندرم کلاین فلتر را میتوان قبل از تولد از طریق آنالیز سایتوژنیک بر روی پرزهای کوریونی یا آمنیوسنتز تشخیص داد. اگر این سندرم قبل از تولد تشخیص داده نشود، بیماری با کاریوتایپ 47,XXY، علائم بالینی وابسته به سن متفاوتی از خود بروز میدهد که تستهای تشخیصی را به چالش میکشد. آنالیز کاریوتایپ لنفوسیتهای خون محیطی، تست سوآب بزاق XCAT-KS، تست FISH و بررسی ریزآرایههای ژن از تستهای بعد از تولد برای تشخیص سندرم کلاین فلتر هستند.
تشخیص سریع و انجام اقدامات مناسب در سندرم کلاین فلتر مهم هستند. کنترل بیماری و درمان علائم باید بر سه مورد متمرکز باشد: هیپوگنادیسم، ژنیکوماستی و مشکلات روانی-اجتماعی. جایگزینی آندروژن (تستوسترون) نقش مهمی در درمان این سندرم دارد.
تیمی متشکل از چند رشتهی تخصصی میتواند بر روی مشکلات گفتاری، تحصیلی، رفتاری و اجتماعی افراد مبتلا به این سندرم کمککننده باشد. فیزیوتراپی برای پسرانی که هیپوتونی (کاهش قدرت عضلانی) عدم تعادل یا تاخیر در تکامل سیستم عصب حرکتی دارند، مناسب است.
تا سال 1996 تصور میشد که مردان مبتلا به سندرم کلاین فلتر، نازا هستند. از آن زمان، با پیشرفت تکنیکهای جراحی و ارتقا تکنولوژی اندامهای جنسی مصنوعی (ART) امکان باروری این مردان بیش از پنجاه درصد افزایش یافته است.
پاتوفیزیولوژی
کروموزوم X حاوی ژنهایی است که در عملکرد اندامهای مختلف مثل بیضهها، تکامل مغز و رشد جسمی نقش حیاتی دارد. وجود یک کروموزوم X اضافی، در نتیجهی یک خطای غیرمستقیم در گامتوژنز یکی از والدین ایجاد میشود که باعث هیپوگنادیسم، ژنیکوماستی و اختلالات رفتاری میگردد.
افزودن یک کروموزوم X یا Y اضافی به کاریوتایپ مردان باعث ایجاد ناهنجاریهای رفتاری و جسمی میشود. هر چه تعداد این کروموزم اضافی بیشتر باشد، اختلالات شناختی و فیزیکی بیشتر تحت تاثیر قرار میگیرد. ناهنجاریهای عضلانی و قلبی میتواند بسیار وخیم باشد. رشد و تکامل اندامهای جنسی به تعداد کروموزوم X حساس است که منجر به دیسژنزی لولههای سمینی فروس و ناباروری میگردد. همچنین عدم رشد و شکل غیرطبیعی اندام تناسلی خارجی در پلیزومی X در مردان بیمار دیده میشود.
شکل اولیهی اختلالات جنسی در مردان مبتلا به سندرم کلاین فلتر، با زیادی میزان هورمونهای جنسی در خون به علت نبود فیدبک منفی توسط هیپوفیز، بروز میکند.
علاوه بر این، ظرفیت ذهنی نیز در حضور کروموزوم اضافی کاهش مییابد. به ازای هر X اضافی، پانزده درجه از میزان IQ افراد کم میشود، اما نتیجه گیری در مورد کاهش تواناییهای ذهنی باید با دقت مورد بررسی قرار گیرد. همهی مراکز اصلی کنترل احساسات، گفتار و هماهنگی رفتاری تحت تاثیر X اضافی هستند.
مطالعهای که توسط وَن ریان و همکارانش انجام گرفته، نشان میدهد که پردازش اطلاعات در مردان با کاریوتایپ 47,XXY به سختی صورت میگیرد که بار اجتماعی بر دوش بیمار میگذارد. به طور مثال، با توجه به پردازش متقاطع که بدون بار اجتماعی است، 17 درصد از افراد در این مطالعه با مشکل مواجه بودند، در حالی که در رابطه با تشخیص چهره (بار اجتماعی متوسط) و حالات صورت (بار اجتماعی بالا)، به ترتیب 26 درصد و 33 درصد از بیماران دچار مشکل بودند. کمبود آندروژن باعث علائم زیر میشود:
کاهش اندازهی اندامهای جنسی/توزیع زنانهی چربیهای بدن
پراکندگی یا عدم وجود موهای صورت، زیربغل، پوبیک و موهای کل بدن
ژنیکوماستی یا بزرگی پستان
کاهش حجم و قدرت عضلانی
کاهش استقامت فیزیکی
بیضه و آلت تناسلی کوچک
کاهش توان جنسی
از دست رفتن عملکرد لولههای سمینیفروس و سلولهای سرتولی و کاهش میزان inhibin B (تنظیم کنندهی میزان FSH)
تغییر محور هیپوتاموس-هیپوفیز گنادی در پسران بالغ
مردان مبتلا به سندرم کلاین فلتر در معرض ابتلا به بیماریهای خودایمنی نظیر دیابت ملیتوس و عوارض ناشی از آن، پوکی استخوان و کاهش تراکم استخوانی، تومورهای پستانی و سلولهای جنسی، لوپوس اریتروماتوز، روماتوئید آرتریت و سندرم شوگرن هستند. مقایسهی ریسک ابتلا به این بیماریها نسبت به خانمهای 46,XX سنجیده میشود.
نتایج آزمایشات خونی مرد مبتلا به این سندرم نشانگر میزان کم هورمون تستوسترون، افزایش هورمون LH و FSH، و افزایش سطح استرادیول است. کاهش تولید تستوسترون در طول عمر فرد بیمار رفته رفته بیشتر میشود، ولی لزوما همهی مردان بیمار دچار هیپوگنادیسم نیستند. هنوز به طور کامل مشخص نیست که عوارض جانبی ناشی از سندرم کلاین فلتر به دلیل هیپوگنادیسم و میزان زیاد استروژن میباشد یا به عملکرد غیرطبیعی ژنهای کروموزوم X مربوط است.
مطالعهی کلوز و همکارانش بر روی پسران مبتلا به این سندرم بیانگرارتباط نزدیکی بین ناهنجاریهای فنوتایپی و کاهش کیفیت زندگی میباشد. تجزیه و تحلیل رگرسیون خطی نشان میدهد که تفاوت 22 درصدی در کیفیت زندگی، بین چهلودو پسر مورد مطالعه وجود دارد.
تحقیقات محققی دانمارکی به نام شاکباک حاکی از آن است که افراد مبتلا به سندرم کلاین فلتر با کاهش کیفیت ذهنی و فیزیکی زندگی مواجه میشوند که مستقیما توسط این سندرم و به طور غیرمستقیم توسط عواملی ایجاد می شود که در مقایسه با گروه کنترل، شامل سطوح پایین درآمد، فعالیت بدنی وعملکرد جنسی است.
اپیدمیولوژی
شیوع
سندرم کلاین فلتر (آناپلوئیدی XXY) رایجترین اختلال کروموزوم جنسی است.
تقریبا یک در 500 یا 600 جنس مذکر با یک کروموزوم اضافی به دنیا میآیند.
شیوع این سندرم پنج تا بیست برابر بیشتر از کسانی است که نسبت به جمعیت مردانه از لحاظ ذهنی مشکل دارند.
حدود دویستوپنجاههزار مرد در آمریکا به سندرم کلاین فلتر مبتلا هستند.
مرگ و میر/عوارض
حدود 40 درصد از بارداریهای کلاین فلتر زنده مانده و به دوران نوزادی میرسند.
به طور کلی، وخامت ناهنجاریهای سوماتیک در کلاین فلتر با تعداد X اضافی متناسب است. عقبماندگی ذهنی و هیپوگنادیسم در بیماران 49,XXXY نسبت به 48XXY بیشتر است.
میزان مرگ و میر در اثر این سندرم تغییری نمیکند.
نژاد
شیوع سندرم کلاین فلتر با نژادهای مختلف ارتباطی ندارد.
جنسیت
سندرم کلاین فلتر نتیجهی اضافه شدن کروموزوم X اضافی به کاریوتایپی با زمینهی XY میباشد، در نتیجه این سندرم فقط در مردان وجود دارد.
سن
این سندرم به ندرت در مردان جوان تشخیص داده میشود. تشخیص کلاین فلتر معمولا در سنین بزرگسالی صورت میگیرد. با این وجود حدود 75 درصد آناپلوئیدیهای کروموزومهای جنسی هرگز تشخیص داده نمیشوند. تهیهی کاریوتایپ بعد از شایع ترین علائم آن، یعنی هیپوگنادیسم و ناباروری انجام میگیرد.
وراثت
سندرم کلاین فلتر ارثی نیست؛ اضافه شدن یک کروموزوم X به هنگام ساخت سلولهای جنسی (تخمک و اسپرم) در یکی از والدین فرد بیمار رخ میدهد. در طول تقسیم سلولی، خطایی غیرمستقیم از توزیع متقارن کروموزومها در سلولهای جنسی جلوگیری میکند. در حالت طبیعی، بعد از تقسیم هر یک از تخمکها، یک نسخه از کروموزوم X و اسپرم یا یک نسخه از کروموزوم X یا Yرا دریافت میکنند. در اثر این جهش، تخمک یا اسپرم با یک نسخهی اضافی از X به وجود میآید.
اگر یک تخمک با دو X با یک اسپرم دارای یک Y، یا یک اسپرم با X وY با تخمک دارای یک X لقاح یابد، جنین حاصل مبتلا به سندرم کلاین فلتر خواهد بود.
حالت موزائیک این بیماری (47,XY/47,XXY) نیز ارثی نیست. این حالت در اثر یک جهش معمولی در تقسیم سلول تخم به وجود میآید. درنتیجه بعضی از سلولهای بدن کاریوتایپ نرمال46,XY و برخی سلولها دارای کاریوتایپ 47,XXY خواهد بود.
علل بیماری
در سال 1959 دانشمندان کشف کردند که سندرم کلاین فلتر، در اثر وجود کروموزوم اضافی X رخ میدهد. وقتی جفت کروموزوم X به طور مساوی تقسیم نشود (طی میوز یک و دو، یا اووژنز و اسپرماتوژنز)، کاریوتایپ 47,XXY ایجاد میشود. خطا در تقسیم سلولهای جنسی پدر یا مادر، حدود 50 درصد در بروز این بیماری موثر است. 75 درصد از خطاهای تقسیم سلول مادری، طی میوز یک اتفاق میافتد که با افزایش سن مادر رابطهی مستقیم دارد. سن بالای پدر نیز احتمالا در رخداد این جهشها تاثیر دارد.
جهشهایی که بعد از لقاح اتفاق میافتد، عامل ایجاد حالت موزائیسم سندرم کلاین فلتر میباشد. مردانی که به این حالت مبتلا هستند، معمولا علائم خاصی از خود نشان نمیدهند و حتی گاهی بیماریشان تشخیص داده نمیشود. ژنهای گیرندهی آندروژن (AR) بر روی کروموزم X قرار گرفتهاند.
- ژن AR شامل توالی پلیمورفیک از کدون (CAG) در اگزون اول است و طول این توالی تکراری CAG به طور معکوس با پاسخ عملکردی گیرنده آندروژن به هورمون آندروژن ارتباط دارد. بنابراین، زنجیرهی کوتاه AR-CAGباعث کارایی بهتر آندروژن میگردد.
- در افراد مبتلا به سندرم کلاین فلتر، کروموزوم X که کوتاهترین توالی AR-CAG را دارد، غیرفعال شده است. این پروسه، غیرفعال سازی نامتوازن کروموزوم X نام دارد.
- بیمارانی که توالی کوتاه AR-CAG دارند، نسبت به درمان، پاسخ امیدوارکنندهای از خود نشان میدهند و در مقایسه با بیمارانی که دارای توالی طولانیتری از AR-CAG هستند، به درجات بالاتری از آموزش و روابط اجتماعی دست پیدا میکنند.
- در مقابل، توالی طولانی AR-CAG باعث افزایش قد و طول دستها، کاهش تراکم استخوان، کاهش حجم بیضهها و ژنیکوماستی میشود.
- غیرفعالسازی نامتوازن کروموزم X که ترجیحا طولانی ترین توالی AR-CAG را فعال میکند، احتمالا با فنوتیپ هیپوگنادیسم در سندرم کلاین فلتر ارتباط دارد و میتواند برخی ناهنجاریهای فیزیکی در بیماران را توضیح دهد.
- در پسران مبتلا به کلاین فلتر، کروموزوم اضافی با منشا پدری، باعث بلوغ دیررس و وجود توالی طولانی CAG از گیرنده ی آندروژن، با تاخیر در فعالسازی محور هیپوفیزی-گنادی همراه است.
- رایجترین کاریوتایپ سندرم کلاین فلتر 47,XXY میباشد که 80 تا 90 درصد موارد بیماری را شامل میشود. فرم موزائیسم (46,XY/47,XXY) در ده درصد موارد بیماری دیده میشود. انواع نادر کاریوتایپهای این سندرم 48,XXYY; 48,XXXY; 49,XXXYY; 49,XXXXY هستند.
- فرم موزائیسم در اثر جهش غیرمستقیم در تقسیم میتوز سلول زیگوت (بعد از لقاح) رخ میدهد که به شکل کاریوتایپهای 46,XY و 47,XXY بروز میکند.
- اشکال دیگر سندرم کلاین فلتر به صورت 48,XXXY; 49,XXXXY; 48,XXYY; 49,XXXYY نیز دیده میشود.
درمان
تشخیص به موقع و انجام اقدامات اولیه در سندرم کلاین فلتر بسیار اهمیت دارد. مراحل درمانی و کنترل بیماری باید بر روی سه مورد تمرکز کند: هیپوگنادیسم، ژنیکوماستی و اختلالات روانی و اجتماعی.
درمان جایگزینی آندروژن
مهمترین بخش درمان، جایگزینی آندروژن یا همان تستوسترون است. مدتها پیش، درمان با تستوسترون هنگام بلوغ، حدودا در دوازده سالگی شروع شده و تا زمانی که سطح سرمی هورمونهای تستوسترون، استرادیول، FSH و LH به مقدار نرمال در سن مورد نظر برسد، مقدار دوز مصرفی افزایش مییابد. در حال حاضر، به علت تغییرات کروموزوم X و Y، نظارت دقیق بر رشد و بلوغ افراد صورت میگیرد تا زمان شروع درمان با تستوسترون را به درستی تعیین کند. برای بعضی از مردان XXY، مشاورهی تغییر جنسیت باید انجام شود.
درمان با آندروژن، کمبود آن را در بدن جبران میکند؛ لذا باعث به وجود آمدن صفات ثانویهی جنسی و نرمال شدن وضعیت فیزیکی بدن میگردد. تزریق مداوم و برنامه ریزی شدهی تستوسترون، باعث افزایش قدرت عضلانی و رشد موها، حجیم شدن عضلات، افزایش میل جنسی، افزایش سایز بیضهها، بالار رفتن اعتمادبهنفس بیمار و جلوگیری از پوکی استخوان میشود. علاوه بر این، اثرات بلند مدت درمان با تستوسترون باعث کاهش ریسک ابتلا به بیماریهای خودایمنی و سرطان پستان میگردد. با این وجود، این پروسه، ناباروری و ژنیکوماستی را درمان نمیکند.
مطالعهای که سامانگو-اسپراس انجام داده، نشان میدهد که هورمون درمانی زودرس (EHT) باعث بهبود رفتارهای اجتماعی در پسران مبتلا به سندرم کلاین فلتر میشود. افراد مورد مطالعه، پسرانی بودند که قبل از تولد با 47XXY تشخیص داده شدهاند. به بیستونه نفر از این پسران، در سه نوبت 25 میلیگرم تستوسترون تزریق شد و با پنجاهوهفت نفر از افراد گروه کنترل که تحت EHT قرار نگرفته بودند، مقایسه شدند. همان طور که با استفاده از پرسشنامه رتبه بندی عملکرد اجرایی، مقیاس پاسخگویی اجتماعی (دومین ویرایش) و چک لیست رفتار کودکان برای سن شش تا هیجده ساله مورد آزمایش قرار گرفتند، محققان تغییرات قابل توجهی در رفتارهای اجتماعی در گروه EHT در مقایسه با گروه کنترل مشاهده کردند. آنها معتقدند که هورمون درمانی، اختلالات تکاملی و رفتاری را در پسران 47,XXY به حداقل میرساند. یک مطالعهی دوسوکور تصادفی توسط راس و همکارانش بر روی پسران مبتلا به سندرم کلاین فلتر پیش از سن بلوغ انجام گرفت. آنها با هورمون درمانی در دوز پایین، بهبود عملکرد بینایی-حرکتی، ترمیم محور هیپوفیزی-گنادی، کاهش افسردگی و اختلالات روانی را گزارش کردند. اگر چه، در عملکرد شناختی و رفتارهای خشونتی و بیش فعالی، تغییر چشمگیری ایجاد نشد.
تیمی متشکل از متخصصان میتواند در گفتار درمانی، بهبود وضعیت تحصیلی، افزایش اعتمادبهنفس و مهارتهای زبانی در کودکان بیمار نقش مهمی ایفا کند. پسران مبتلا به سندرم کلاین فلتر باید مورد ارزیابی روانشناختی جامع (IEP) قرار بگیرند تا نقاط ضعف و قوت خود را کشف کنند. اطلاعاتی که از این ارزیابیها بدست میآید، در برنامهریزی منابع آموزشی مناسب و قرار دادن بیماران در کلاس درس مفید باشد.
درمان فیزیکی و شغلی
فیزیوتراپی برای بیمارانی که از هیپوتونی یا عدم هماهنگی حرکات بدن رنج میبرند و یا به دیسپاراکسی مبتلا هستند، به کار میرود.
درمان ناباروری
تا سال 1996 مردان مبتلا به سندرم کلاین فلتر نابارور تصور میشدند. از آن زمان، پیشرفت تکنیکهای جراحی و ارتقا تکنولوژی اندامهای جنسی مصنوعی (ART) امکان باروری را در 50 درصد مردان بیمار ایجاد کرده است. موفقیتی در این زمینه، از ترکیب استخراج اسپرم میکروسکوپیک بیضه (TESE) و اسپرم تازه بازیافت شده در روش IVFحاصل شد. TESE روشی است که در آن نمونهی کوچکی از بافت بیضه را برداشت کرده و اسپرمهای سالم را برای تزریق اسپرم داخل سیتوپلاسمی (ICSI) استخراج میکنند. این روش به مردان امکان میدهد تا صاحب فرزند شوند. مطالعهای که بر روی چهل ودو مرد مبتلا به سندرم کلاین فلتر انجام شد، نشان میدهد که به ازای هر نمونه برداری از بیضه، 72 درصد احتمال استخراج اسپرم بارور وجود دارد. بنابراین، تکنیکهای TESE و ICSI برای باروری مردانی که دچار آزواسپرمی هستند، به کار میرود. بعضی از مردان بیمار، دارای اسپرم بارور در مایع منی خود هستند که باروری طبیعی آنان را ممکن میسازد.
مشاورهی ژنتیک
اتیولوژی 47,XXY وآناپلوئیدیهای کروموزوم جنسی به خطاهای غیرمستقیم تقسیم سلولی مربوط است. خطر بروز این جهشها وراثتی نیست، از این رو، در یک خانواده که بیمار مبتلا به کلاین فلتر دارند، احتمال بروز دوبارهی جهش بیشتر نمیشود. پزشکان و مشاوران ژنتیک باید والدینِ کودکان مبتلا به سندرم کلاین فلتر را آگاه کنند تا این کودکان به درستی تا سن بلوغ تحت نظارت پزشک قرار گیرند. بهترین زمان برای در جریان گذاشتن فرد بیمار، سنین بزرگسالی است تا بتواند با شرایط خود کنار بیاید.
مشاورهی ژنتیک باروری
اسپرمهایی که از بیماران غیرموزائیک با کاریوتایپ 47,XXY استخراج شده بود، با موفقیت در فرایند تولیدمثل مورد استفاده قرار گرفت. با این وجود، علت اصلی وقوع جهش در تقسیم میوز بیماران غیرموزائیک با کاریوتایپ 47,XXY هنوز مشخص نیست.
موزائیسم را نمیتوان در یک بیمار غیرموزائیک تایید کرد. وجود یک سری سلول بنیادی XY در بیضه میتواند تولید سلولهای هاپلوئید سالم را در بیماران ظاهرا غیرموزائیک توجیه کند. با این اوصاف، تهیهی کاریوتایپ از لنفوسیتهای محیطی نمیتواند ژنوم سلولهای بیضه و وجود یا عدم وجور اسپرماتوژنز را پیشبینی کند. ICSIریسک آنومالیهای کروموزومی را در نسل بعد افزایش میدهد. IVF نیز باعث رخ دادن جهشهای کروموزومی جدید، خصوصا در کروموزمهای جنسی میشود.
مشاورهی ژنتیک باروری برای بیماران 47,XXY دشوار است. بعضی از محققان پیشنهاد میکنند که بعد از انجام ICSI باید کاریوتایپ زیگوت را قبل از جایگزینی در رحم (PGD) بررسی کرد تا جهشهای جدید تشخیص داده شود.
احتمال مبتلا شدن نسلهای بعد به سندرم کلاین فلتر در بیماران 47,XXY، بسیار پایین است که این ریسک، کروموزمهای جنسی و آناپلوئیدی اتوزومال را شامل میشود. مشاورهی ژنتیکی باید با اطمینان انجام شده و مدیریت حاملگی با احتیاط ادامه یابد.