هیپرپلازی مادرزادی آدرنال چیست؟
اصطلاح هیپرپلازی مادرزادی آدرنال (CAH) بر گروهی از اختلالات اتوزومال مغلوب دلالت میکند که هر کدام از آنها شامل یک نقص در یک آنزیم دخیل در سنتز کورتیزول، آلدوسترون یا هر دوی این هورمونها میباشد.
پاتوفیزیولوژی هیپرپلازی مادرزادی آدرنال
تظاهرات بالینی هر شکلی از هایپرپلازی مادرزادی آدرنال وابسته به درجهی نقص کورتیزول یا آلدوسترون میباشد. در برخی موارد، این تظاهرات بازتاب تجمع پیشساز هورمونهای آدرنوکورتیکال است. وقتی این پیشسازها با غلظتهای بیشتر از غلظت فیزیولوژیک تجمع یابند، منجر به تولید بیش از حد آندروژن با اثرات ویریلیزاسیون میشوند، یا بهخاطر خصوصیات مینرالوکورتیکوئیدی، موجب احتباس سدیم و پرفشار خون میگردند. فنوتیپ بیمار به درجه یا نوع حذف یا جهش ژنی و نقص آنزیم استروئیدوژنیک در نتیجهی آن، بستگی دارد. این آنزیمها و ژنها مرتبطشان و در تصویر زیر نشان داده شده است.

دو رونوشت از ژن مختل برای وقوع بیمار لازم است، و همهی جهشها و حذفهای نسبی منجر به بیمار نمیشوند. فنوتیپ بیماری میتواند از بدون علائم بالینی (هیپرپلازی آدرنال occult or cryptic) تا شکل خفیفی از یک بیماری در سنین نوجوانی یا بزرگسالی (هیپرپلازی آدرنال غیرکلاسیک) تا بیماری شدید منتج به نارسایی آدرنال در نوزادی با یا بدون ویریلیزاسیون و از دست دادن نمک (هیپرپلازی آدرنال کلاسیک) متفاوت باشد. شایعترین شکل هیپرپلازی آدرنال (به دلیل نقص در فعالیت 21-هیدروکسیلاز) از لحاظ بالینی به سه فنوتیپ تقسیمبندی میشود: با از دست دادن نمک، ویریلیزاسیون ساده، و غیرکلاسیک.
CYP21A ژنی است که 21-هیدروکسیلاز را کد میکند، CYP11B1 11-بتا-هیدروکسیلاز، و CYP17 17-آلفا-هیدروکسیلاز را کد میکنند. بسیاری از آنزیمها دخیل در سنتز کورتیزول و آلدوسترون از پروتئینهای سیتوکروم P450 (CYP) هستند.
تناوب
ایالات متحده
شایعترین شکل هیپرپلازی مادرزادی آدرنال بهخاطر جهش یا حذف CYP21A است که منجر به نقص 21-هیدروکسیلاز میشود. این نقص بیش از 90 درصد موارد هیپرپلازی آدرنال را شامل میگردد. جهشها یا حذفهای نسبی که CYP21A را تحت تأثیر قرار میدهند شایع اند؛ به طوری که تخمین زده میشود 1 نفر از هر 3 نفر در برخی جمعیتهای خاص (مثل یهودیان اشکنازی) تا 1 نفر از هر 7 نفر در شهر نیویورک آنها را داشته باشند. شیوع تخمینی این جهشها 1 در 60 نفر در جمعیت عمومی تخمین زده میشود.
هیپرپلازی کلاسیک آدرنال شیوع کلی 1 در هر 16,000 نفر دارد؛ اگرچه در جمعیتهای منتخب (مثل یوپیک آلاسکا)، شیوع به اندازهی 1 از 400 نفر بالا میرود. هیپرپلازی مادرزادی آدرنال به علت نقص در 11-بتا-هیدروکسیلاز شامل 5-8 درصد از تمام موارد هیپرپلازی مادرزادی آدرنال میشود.
بینالمللی
هیپرپلازی مادرزادی آدرنال به علت نقص 21-هیدروکسیلاز در تمام جمعیتها وجود دارد. نقص 11-بتا-هیدروکسیلاز در مراکشیها یا دودمان یهودیان ایرانی شایعتر میباشد.
مرگ و میر
مرگ و میر اشکال مختلف هیپرپلازی آدرنال بهتر از هر شکل دیگری با فهم مسیر استروئیدوژنیک مورد استفاده در غدد آدرنال و گنادها درک میشود که در پائین نشان داده شده است.
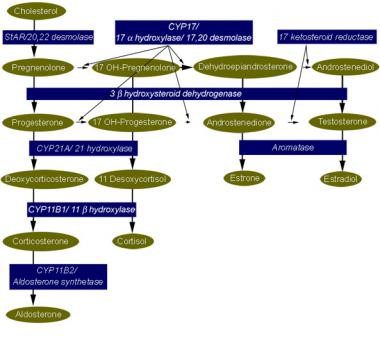
مسیر استروئیدوژنیک برای سنتز کورتیزول، آلدوسترون، و استروئید جنسی. جهش یا حذف هر کدام از ژنهایی که آنزیمهای دخیل در سنتز کورتیزول و آلدوسترون را کد میکنند، موجب هیپرپلازی مادرزادی آدرنال میگردد. فنوتیپ خاص که ایجاد میشود به جنسیت فرد، مرحلهی انسداد در مسیر سنتز، و شدت حذف یا جهش ژنتیکی بستگی دارد.
فنوتیپ بالینی را میتوان از بررسی نقص آنزیم، تجمع پیشساز هورمونها، فرآوردههای این پیشسازها در صورت ناکارآمدی مسیر سنتز، و اثر فیزیولوژیک این هورمونها درک کرد.
مطالعهای از هالپر و همکارانش روی 42 کودک با هیپرپلازی مادرزادی آدرنال گزارش کرد که تراکم استخوانی کلی در این کودکان نسبت به همسالان شاهدشان کمتر میباشد (به ترتیب 0.81 g/cm2 در برابر 1.27 g/cm2). اگرچه، تفاوت قابل ملاحظهای در ترکیب بدن، چربی احشایی و نسبت آندروئید به ژنیکوئید بین این دو گروه پیدا نشد.
مطالعهای از طرف یانگ و وایت نشان داد که در کودکان مبتلا به هیپرپلازی مادرزادی آدرنال بر اثر نقص 21-هیدروکسیلاز با فرم از دست دهندهی نمک، خطر بستری پس از تشخیص در بیماران زیر 2 سال بیشتر است (احتمالاً بهخاطر آسیبپذیری بیشتر نسبت به عفونتهای ویروسی و توانایی پائینتر در سازگاری با استرس و دهیدراسیون) و آنهایی که به دوز روزانهی بالاتری از فلودروکورتیزون نیاز دارند (چون این بیماران احتمالاً بیماریهای شدیدتری دارند). مطالعهی مذکور همچنین به این نتیجه رسید که کودکانی با بیمهی noncommercial بیشتر احتمال دارد بستری شوند؛ چون احتمالاً با موانع اجتماعی بیشتری در راه درمان روبهرو میشوند.
اشکال شدید هیپرپلازی مادرزادی آدرنال اگر شناسایی و درمان نشوند به طور بالقوهای کشنده هستند؛ چون نقص شدید کورتیزول و آلدوسترون موجب از دست دادن نمک، هیپوناترمی، هیپرکالمی، دهیدراسیون، و هیپوتانسیون میگردد.
اپیدمیولوژی
نژاد
هیپرپلازی مادرزادی آدرنال در بین مردم تمام نژادها شیوع دارد. هیپرپلازی مادرزادی آدرنال ثانویه به جهشها و حذفهای CYP21A1 به طور ویژهای در بین اسکیموهای یوپیک شایع میباشد.
جنسیت
چون تمام فرمهای هیپرپلازی مادرزادی آدرنال اختلالات اتوزومال مغلوب هستند، با احتمال برابری در هر دو جنس به وجود میآید. اگرچه، چون پیشسازهای تجمعیافتهی هورمونها یا سنتز مختل شدهی تستوسترون روی تمایز جنسی تأثیر میگذارد، عواقبت فنوتیپی جهشها و حذفهای یک ژن خاص بین جنسیتها با هم فرق میکند.
سن
هیپرپلازی مادرزادی آدرنال عموماً در هنگام تولد یا اوایل کودکی تشخیص داده میشود؛ چون ابهام جنسی، از دست رفتن نمک یا ویریلیزاسیون زودهنگام واضح است. هیپرپلازی غیرکلاسیک آدرنال در کل، هنگام بلوغ یا پس از آن بهخاطر الیگومنوره یا علائم ویریلیزاسیون در جنس مؤنث تشخیص داده میشود.
پیشآگهی
با درمانهای طبی و جراحی کافی، پیشآگهی خوب است. اگرچه، برطرف کردن مشکلات روانی که معمولاً بهخاطر ناهنجاری ژنیتال به همراه بعضی از فرمهای هیپرپلازی مادرزادی آدرنال است، شایع میباشد.
کوتاهی قد و ناباروری شایع است.
اگر نوزاد از همان اول دختر شناخته شود، و پشتیبانی طبی و جراحی کافی فراهم گردد، و اگر خانواده (و در نهایت خود بیمار) در مورد بیماری خوب تفهیم شود، هویت جنسی در دخترانی که مبتلا به هیپرپلازی ویریلیزه کننده آدرنال هستند، مؤنث شکل میگیرد.
دخترانی که مبتلا به هیپرپلازی ویریلیزه کننده آدرنال هستند شاید تمایلات مردانهی بیشتری داشته باشند.
زنان مبتلا به هیپرپلازی آدرنال نرخ باروری پائینتری دارند، اما با کنترل متابولیک خوب، باروری امکانناپذیر هم نیست.
اگر دوزهای کافی گلوکوکورتیکوئید در زمان بیماری، تروما یا جراحی برای این بیماران فراهم نگردد، مرگ زودهنگام دور از انتظار نیست.
علل هیپرپلازی مادرزادی آدرنال
نقایصی که موجب هیپرپلازی مادرزادی آدرنال میشوند اختلالات اتوزومال مغلوب به دلیل فعالیت ناقص یک پروتئین دخیل در سنتز کورتیزول، آلدوسترون یا هر دوی آنهاست.
در برخی موارد، این اختلال به دلیل یک جهش یا حذف ژنی است که پروتئین مورد نظر را کد میکند. وقتی هر دو ژن حامل جهش یا حذف یکسان باشند، این عارضه هموزیگوت است. در کل، شدت بالینی حداقل آللهای تحت تأثیر قرار گرفته را بازتاب میکند. ناقلین یا هتروزیگوتهایی تنها یک ژن ناهنجار را حمل میکنند، بدون علائم هستند.
بسیاری از ژنهای دخیل در سنتز کورتیزول و آلدوسترون پروتئینهای CYP را کد میکنند. ژنی که بیشتر از همه در این میان مورد مطالعه قرار گرفته، ژن 21-هیدروکسیلاز (CYP21, CYP21A) میباشد. ژن 21-هیدروکسیلاز باند کروموزومی 6p21.3 در میان سایر ژنهایی است که تایپهای آنتیژن لکوسیت انسانی (HLA) را کد میکنند. ژن 21-هیدروکسیلاز ژن کاذبی (CYP21P) به فاصلهی 30 کیلوباز از CYP21 دارد که از لحاظ ساختار 98 درصد با CYP21A همولوگ میباشد؛ اگرچه به علت تفاوتهای جزئی به صورت غیرفعال درآمده است. اعتقاد بر این است که مجاورت CYP21P با CYP21A، ژن CYP21A را در معرض کراساوو با CYP21P در میوز قرار داده است که باعث میشود عملکرد ژنتیکی از بین برود.
سایر نقایص به علت جهشها و حذفهای ژنی اتفاق میافتند. در بین ناهنجاریهای CYP21A، تقریباً 95 درصد احتمالاً به دلیل نوترکیبیها با CYP21P، 20 درصد ناشی از حذفها، و 70 درصد جهشهای نقطهای هستند. فنوتیپ وابسته به عملکرد ژنی است که کمترین آسیب را دیده است، نه ژنی که بیشترین آسیب را دیده؛ چون اولی سطح فعالیت آنزیم را تعیین میکند. در کل، ارتباطات ژنوتیپ-فنوتیپ به جز استثنائاتی که رخ میدهد قوی است. چون ترشح آلدوسترون تقریباً 1000 برابر کمتر از کورتیزول است، فعالیت آنزیمی مورد نیاز آلدوسترون کمتر از فعالیت آنزیمی مورد نیاز کورتیزول میباشد. بنابراین، تنها بیمارانی که شدیدترین فقدان عملکرد را در CYP21A دارند از دست رفتن نمک را تجربه خواهند کرد.
ژن 11-بتا-هیدروکسیلاز (CYP11B1) روی باند کروموزومی 8q21 قرار دارد. CYP11B1 هیچ ژن کاذبی ندارد و هیچ ارتباطی HLA نیز یافت نشده است.
CYP11B1 تبدیل 11-دئوکسیکورتیزول را در مسیر گلوکوکورتیکوئید به کورتیزول و تبدیل دئوکسیکورتیکوسترون را به کورتیکوسترون در مسیر مینرالوکورتیکوئید کاتالیز میکند. ژنی در همسایگی CYP11B2یا همان آلدوسترون سنتتاز را کد میکند که تبدیل کورتیکوسترون به آلدوسترون را در زونا گلومرولوزا کاتالیز مینماید. جهشها و حذفهای ژن CYP11B2 باعث کاهش سنتز آلدوسترون میگردد. بنابراین افرادی که نقص CYP11B2 دارند دچار هیپوناترمی، هیپرکالمی، و دهیدراسیون میشوند.تمایز جنسی بهخاطر سنتز نرمال استروئیدهای جنسی و کورتیزول اتفاق میافتد. ژنهای CYP11B1 و CYP11B2 95 درصد تشابه ژنی در توالیهای کدکننده دارند. با این وجود، تبدیل ژنی از چلیپایگی کروموزومی در میوز به نظر نمیآید نقش مهمی در جهشها و حذفهایی داشته باشد که ژنها را به حالت غیرفعال درمیآورند.
دو فرم بافتی 3-بتا-هیدروکسیاستروئید دهیدروژناز توصیف شده است. تایپ I اکثراً در آدرنال و گناد تولید میشود، برعکس، تایپ II اکثراً در جفت و کبد فعالیت میکند. هر دوی این ژنها در باند کروموزومی 1p13 قرار دارند. فرم کلاسیک نقص 3-بتا-هیدروکسیاستروئید دهیدروژناز از جهشها یا حذفها در این ژنها برای فرم آدرنال آنزیم ایجاد میگردد.
به نظر میآید برخی از بیماران فرمهای غیرکلاسیک این بیماری را داشته باشند؛ چون شواهدی با علائم و نشانههای ویریلیزاسیون، مثل هیرسوتیسم، الیگومنوره و ناباروری دیده شده است. مطالعات آزمایشگاهی نسبتهای پیشساز-فرآورده کمی غیرعادی (مثل افزایش نسبت 17-هیدروکسیپرگننولون به 17-هیدروکسیپروژسترون و دیهیدرواپیآندروسترون به آندروستروندیون) را نشان دادهاند. این بیماران جهش یا حذفی در هیچ کدام از ژنهای کدکنندهی 3-بتا-هیدروکسیاستروئید دهیدروژناز ندارند. اساس مولکولی این اختلال ناشناخته مانده است. یافتههای هورمونی و بالینی این عارضه و بیماری تخمدان پلیکیستیک به طور قابل ملاحظهای همپوشانی دارند. برخی از بیماران از سرکوب استروئیدوژنز آدرنال با دگزامتازون سود میبرند.
فعالیت 17-آلفا-هیدروکسیلاز و 17,20-دسمولاز به نظر میرسد زیر سر پروتئینی واحد (CYP17) با محل فعالیت آنزیمی متفاوت باشد.
بعضی از بیماران با هیپرپلازی آدرنال لیپوئید که اساساً فکر میکنیم به دلیل نقص فعالیت آنزیمی در کلیواژ زنجیره جانبی CYP450 باشد، جهشهایی در ژنی داشتهاند که StAR را کد میکند. به نظر میآید این پروتئین در انتقال کلسترول از عرض غشای میتوکندری دخیل است، جایی که CYP450 scc میتواند آنجا عمل کند. این آنزیم کلسترول را به پرگننولون تبدیل میکند که سپس در بافتهای استروئیدوژنیک مختلف به کورتیزول، آلدوسترون یا استروئیدهای جنسی پردازش میگردد. بنابراین، نقص StAR به یک وضعیت نقص کلی استروئیدی میانجامد. بیماران 46 XY میتوانند اندامهای جنسی خارجی زنانه داشته باشند، و بیماران 46 XX اندامهای جنسی نرمال زنانه خواهند داشت. هر دو نارسایی آدرنال با ظهور زودهنگام از شیرخوارگی تا شش ماهگی دارند.
یکی از مشاهدات کنجکاوانه این است که زنان مبتلا به این اختلال که با جایگزینی زودهنگام گلوکوکورتیکوئیدها و مینرالوکورتیکوئیدها زنده ماندهاند، هنگام بلوغ پستانهایشان رشد کرده و منس خودبهخودی غیرتخمدانی داشتهاند.
محققان این گونه نتیجهگیری کردهاند که انباشت استرهای کلسترول در سلولهای استروئیدوژنیک، که ناشی از نقص StAR است، نهایتاً برای این سلولها سمی میشود. بر اساس این نظریه، برخی از عملکردهای تخمدانی حفظ میشود؛ چون استروئیدوژنز تخمدان تا زمان بلوغ شروع نمیشود و پس از آن نیز استروئیدوژنز تنها در یک فولیکول در یک زمان اتفاق میافتد؛ به همین دلیل برخی عملکردها حفظ میگردد.
جهشها در ژنی که CYP اکسیدوردوکتاز را کد میکند اخیراً در ایجاد نقص در چندین آنزیم دخیل در استروئیدوژنز مقصر شناخته شده است. CYP اکسیدوردوکتاز انتقال الکترون را از فرم کاهش یافتهی نیکوتینآمید آدنین دینوکلئوتید فسفات (NADPH) به 21-هیدروکسیلاز و 17-هیدروکسیلاز تسهیل مینماید که در استروئیدوژنز الزامی است. برخی افراد دارای این جهشها کرانیوسینوستوز و ناهنجاریهای اسکلتی به نام سندرم انتلی-بیکسلر را نشان میدهند. اگرچه، جهشهایی در گیرندهی فاکتور رشد فیبروبلاست 2 نیز میتواند سبب ایجاد تصویر فنوتیپی سندرم انتلی-بیکسلر بدون مشکلات استروئیدوژنز گردد.
درمان هیپرپلازی مادرزادی آدرنال
وقتی تشخیص اندام جنسی مبهم در نوزادی داده شد، بایستی برای مشاهدهی علائم و نشانههای از دست رفتن نمک با دقت تحت نظر باشد. سرنخهای بالینی شامل کاهش وزن غیرعادی یا فقدان افزایش وزن مورد نظر میباشد. اختلالات الکترولیتی در کل از چند روز تا 3 هفته زمان نیاز دارند تا خود را نشان دهند؛ چون جفت الکترولیتهای جنین را در رحم ثابت نگه میدارد. در فرمهای خفیف از دست رفتن نمک در هیپرپلازی آدرنال، شاید تا وقتی یک بیماری نوزاد را در شرایط استرسی قرار نداده، از دست رفتن نمک معلوم نگردد.
بیمارانی با دهیدراسیون، هیپوناترمی، یا هیپرکالمی و یک شکل احتمالی از دست دادن نمک در هیپرپلازی آدرنال، با یک یک بولوس داخل وریدی از محلول سدیم کلرید ایزوتونیک (20 mL/kg or 450 mL/m2) در ساعت اول در صورت نیاز دریافت نمایند تا حجم داخل عروقی آنها و فشار خونشان بازیابی شود.
اگر فشار خون پائین بماند این دوز میتواند تکرار شود.
اگر بیمار هیپوگلیسمیک است، دکستروز باید تجویز شود و بایستی پس از دوز بولوس برای پیشگیری از هیپوگلیسمی به داخل مایع رهیدراسیون نیز اضافه گردد.
پس از نمونهگیری به منظور اندازهگیری غلظت الکترولیت، قند خون، کورتیزول، آلدوسترون و 17-هیدروکسیپروژسترون، باید درمان با گلوکوکورتیکوئیدها بر اساس نارسایی مورد تردید آدرنال آغاز شود. نباید تا دریافت پاسخ آزمایشها درمان را عقب انداخت؛ چون میتواند جان بیمار را به خطر بیاندازد.
پس از تثبیت شرایط بیمار، همهی بیماران هیپرپلازی آدرنال را باید با درمان جایگزینی طولانیمدت گلوکوکورتیکوئید و آلدوسترون، بسته به آنزیم دخیل و تحت تأثیر قرار گرفتن کورتیوزل و/یا آلدوسترون درمان کرد.
اپروچ دیگری که هماکنون در دست بررسی است، استفادهی ترکیبی از گلوکوکورتیکوئید (برای سرکوب ACTH و تولید آندروژن آدرنال)، مینرالوکورتیکوئید (برای کاهش غلظت آنژیوتانسین II)، مهارکنندهی آروماتاز (برای کند کردن بلوغ استخوانی)، و فلوتامید (یک آندروژن بلاکر به منظور کاهش ویریلیزاسیون) میباشد.
بعضی از بیماران با بلوغ precocious روبهرو میشوند که بیش از پیش موجب سرکوب رشد قدی آنها میشود. سرکوب بلوغ با آگونیستهای هورمون آزادکنندهی گنادوتروپین طولانی اثر (GnRH) همزمان با تحریک رشد توسط هورمون رشد میتواند قد بیمار را تا حدودی بهبود بخشد.
راهنمای بالینی 2010 انجمن اندوکرین موارد زیر را خاطرنشان شده است:
- درمان پیش از تولد برای هیپرپلازی مادرزادی آدرنال باید آزمایشی در نظر گرفته شود.
- برای جلوگیری از سندرم کوشینگ، تیتراسیون گلوکوکورتیکوئید تراپی باید با دقت انجام شود.
- جایگزینی مینرالوکورتیکوئید توصیه میشود. در شیرخواران، جایگزینی مینرالوکورتیکوئید و مکملیاری سدیم توصیه میگردد.
- استفاده از عواملی برای به تأخیر انداختن بلوغ و تقویت رشد آزمایشی هستند.
- حمایت روانی باید برای بیمارانی که نیازمند هستند در نظر گرفته شود.
- دارودرمانی در بارداری و بیماران هیپرپلازی مادرزادی آدرنال با علائم، باید با احتیاط انجام شود.